Increasing Resolution in Live Cell Microscopy by Structured Illumination (SIM)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
- -
- stimulated emission depletion (STED) microscopy [6], where the outer regions of an illuminated spot are depleted by stimulated emission within the focus of a second donut-shaped laser beam upon optical excitation in a laser scanning microscope. While a fluorescent spot may thus be confined to 70 nm or less, the irradiance exceeds that of a conventional fluorescence microscope by a factor of 104–105, so at the moment live-cell imaging does not appear to be possible, although various approaches to reduce the light exposure considerably are being pursued. Those methods include reduction of state transition cycles (REScue) [7], adaptive-illumination STED nanoscopy [8] or MINFLUX technology with tracking of single molecules in the center of a donut-shaped laser beam [9];
- -
- super-localization microscopy of single molecules located within a thin illuminated layer of a sample. If a single molecule is detected n times, its localization can be determined with precision, Δx = Δx0/√n with Δx0 ≈ 200 nm resulting from the Rayleigh criterion. Therefore, a precision of localization Δx = 20 nm results when n = 100 and Δx = 10 nm when n = 400. Methods based on super-localization microscopy include stochastic optical reconstruction microscopy (STORM), photoactivation localization microscopy (PALM) and related techniques [10,11,12]. However, the irradiance needed for single-molecule experiments is still 500–1000 times higher than in conventional fluorescence microscopy, such that cells are expected to survive only a short time.
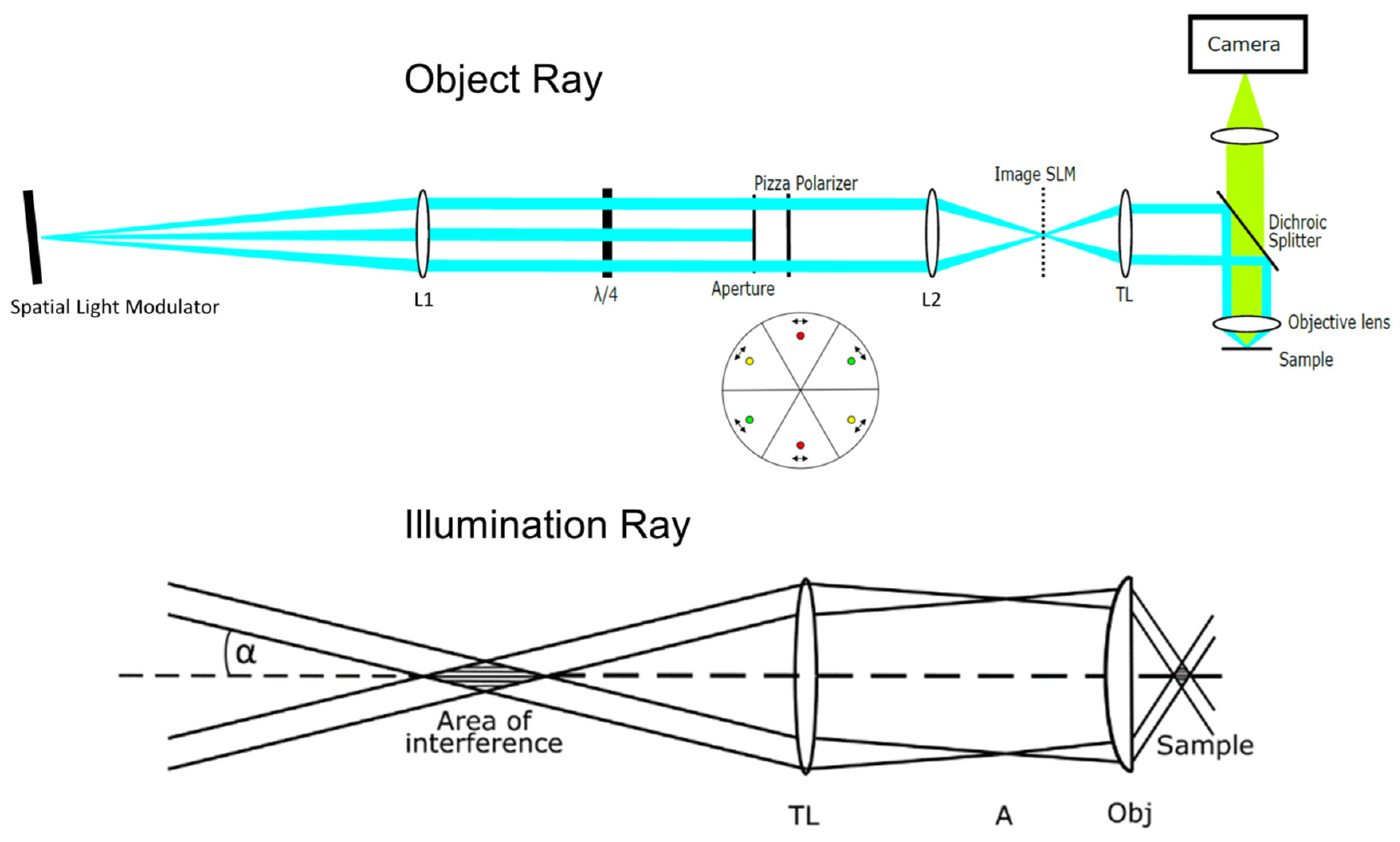
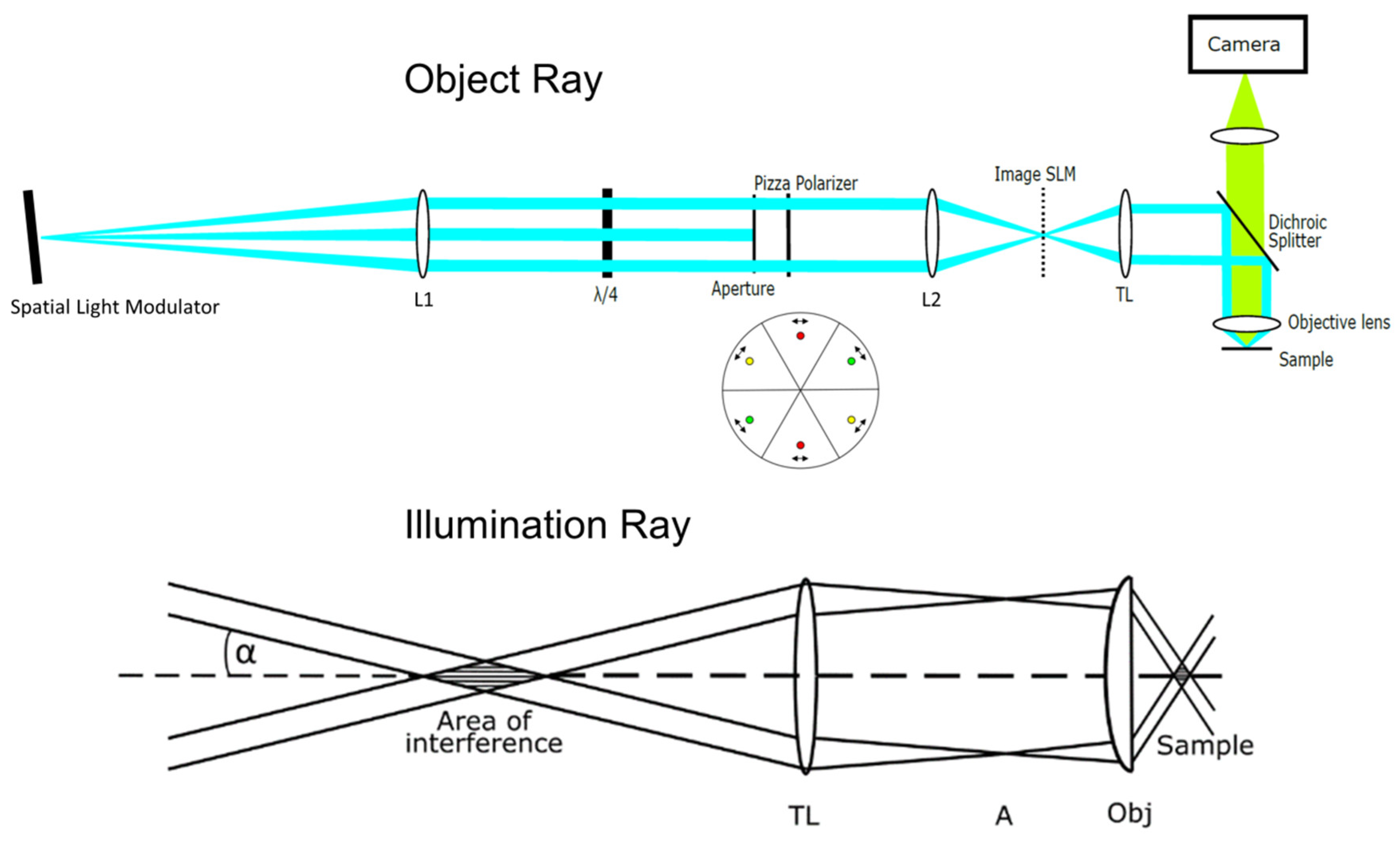
2. Materials and Methods
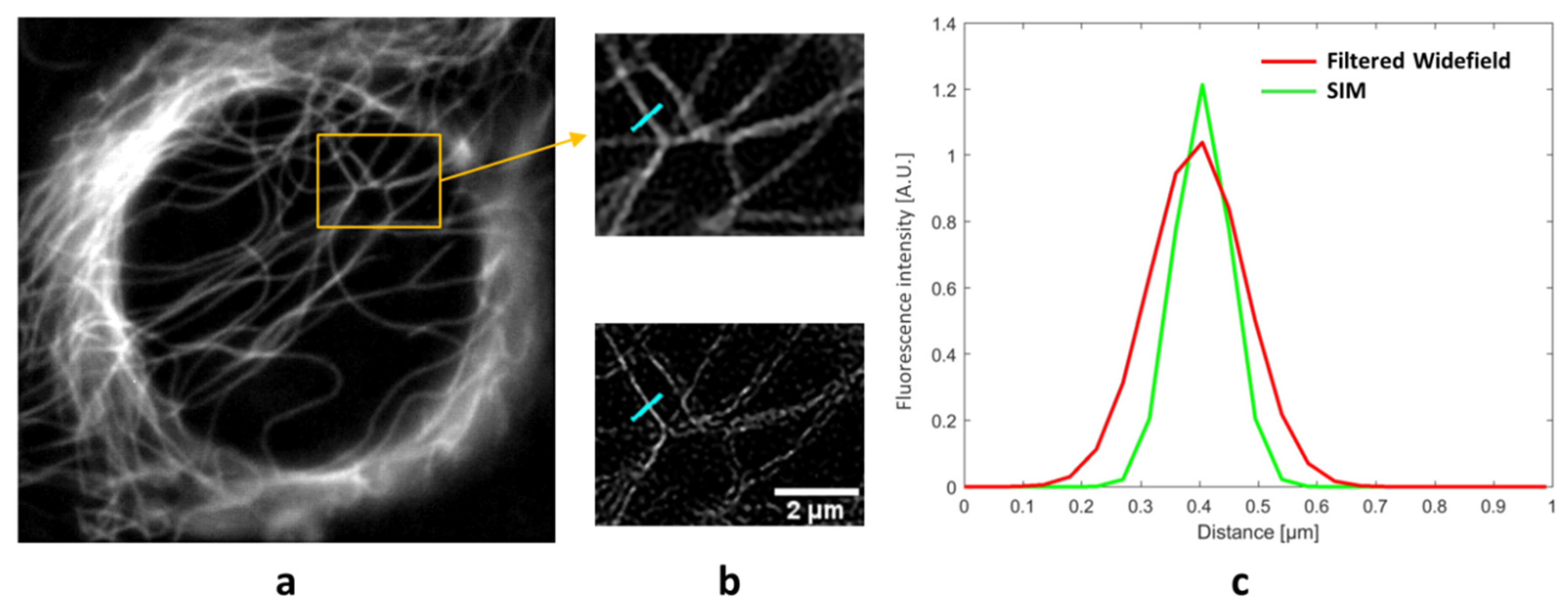
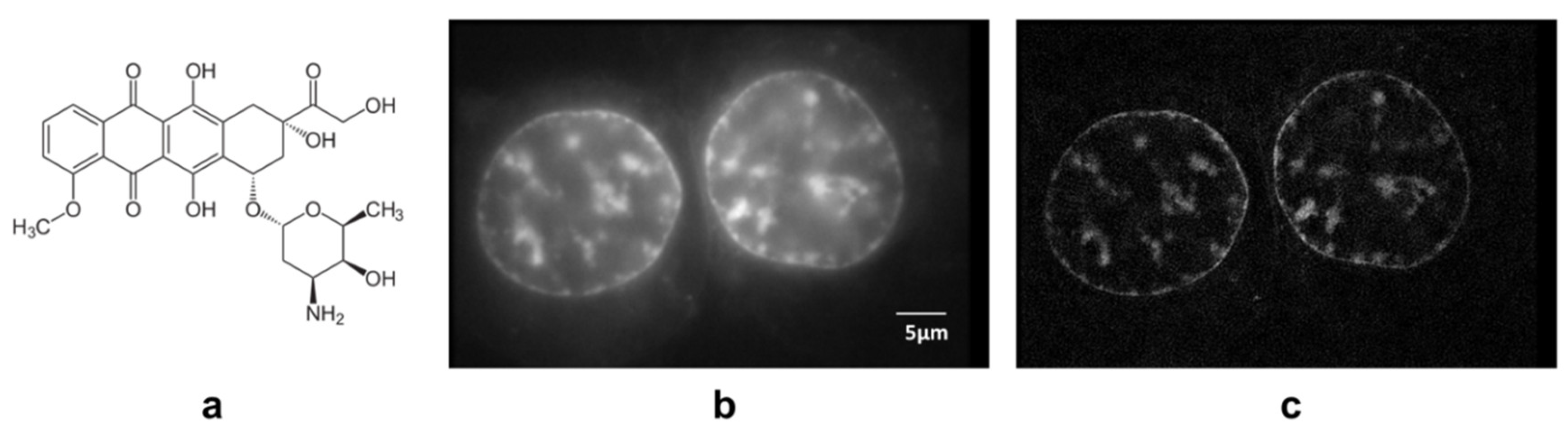
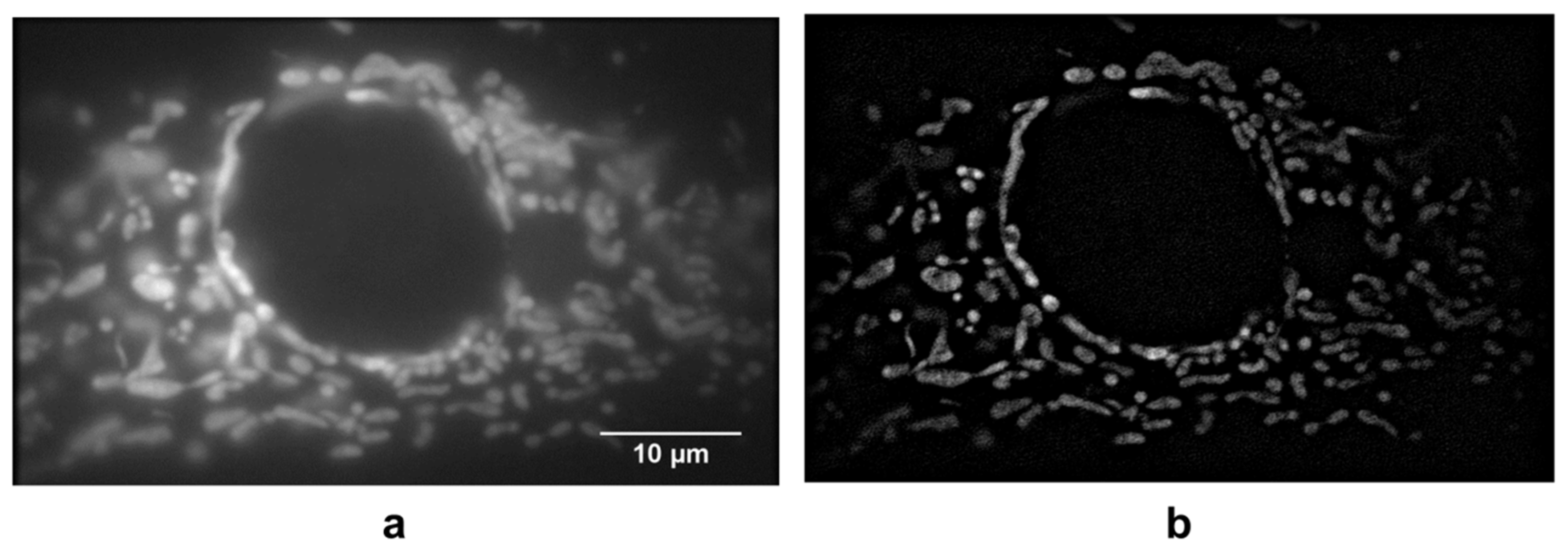
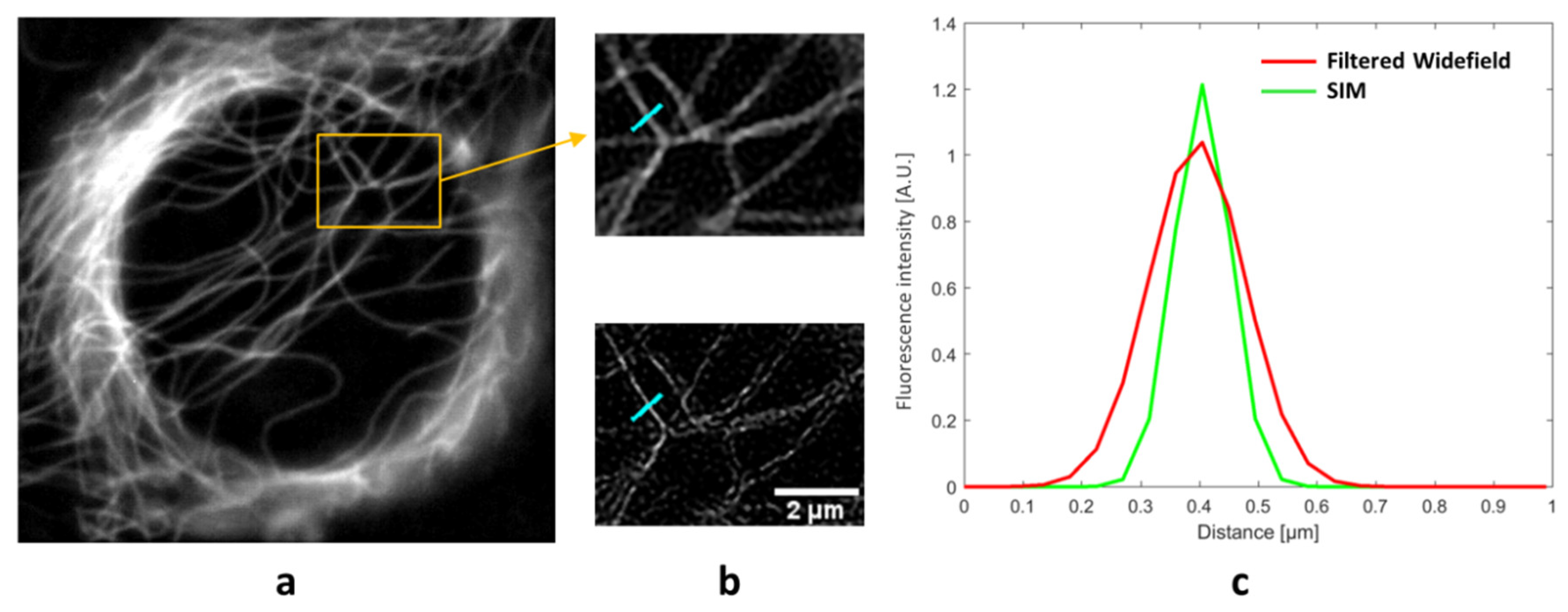
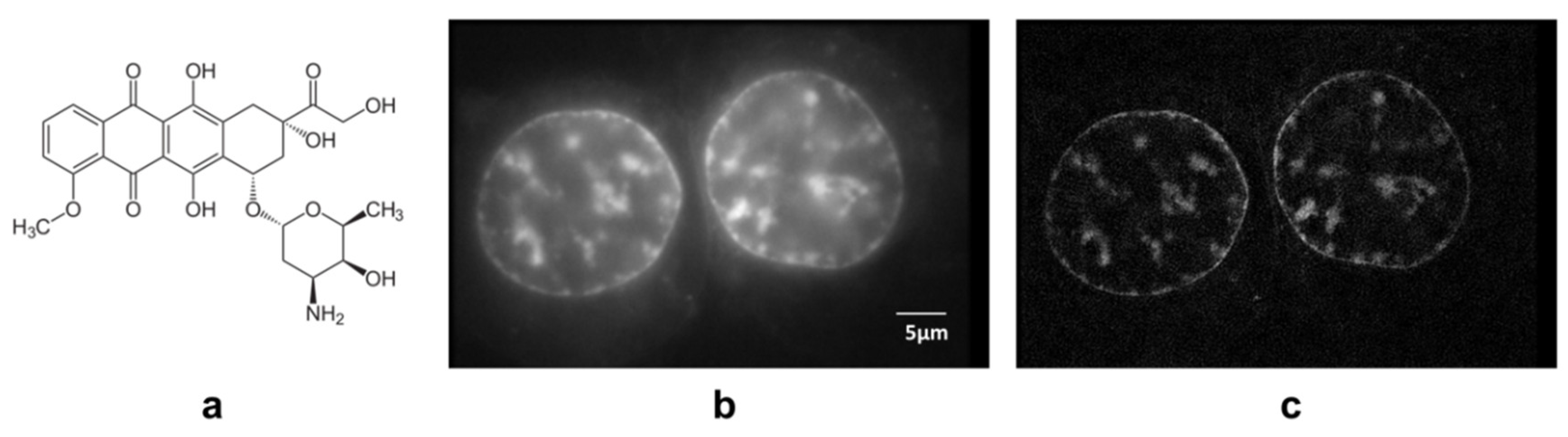
3. Results
4. Discussion
- -
- -
- it is comparably simple, without any moving components, using a commercially available (inverted) microscope;
- -
- microscope lenses of various magnification and aperture can be used. In the present case a 40×/1.30 objective lens permits obtaining a comparably large field of illumination for cell-based experiments, but requires cameras with more than 250 pixels per millimeter (e.g., ProgRes cameras by Jenoptik GmbH or an Orca Flash camera set by Hamamatsu Photonics) to image structures of about 100 nm size; and,
- -
- it has the potential for imaging in larger depths of a sample (“deep view imaging”). This potential can, in principle, even be used for in combination with further super-resolution methods, e.g., STED microscopy where SIM may generate a three-dimensional depletion pattern, permitting axial super-resolution STED with moderate light exposure [31].
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pawley, J. Handbook of Biological Confocal Microscopy, 3rd ed.; Springer: Boston, MA, USA, 1990. [Google Scholar] [CrossRef]
- Webb, R.H. Confocal optical microscopy. Rep. Prog. Phys. 1996, 59, 427–471. [Google Scholar] [CrossRef]
- Pampaloni, F.; Chang, B.-J.; Stelzer, E.H.K. Light sheet-based fluorescence microscopy (LSFM) for the quantitative imaging of cells and tissue. Cell Tissue Res. 2015, 362, 265–277. [Google Scholar] [CrossRef]
- Santi, P.A. Light sheet fluorescence microscopy: A review. J. Histochem. Cytochem. 2011, 59, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Bruns, T.; Bauer, M.; Bruns, S.; Meyer, H.; Kubin, D.; Schneckenburger, H. Miniaturized modules for light sheet microscopy with low chromatic aberration. J. Microsc. 2016, 264, 261–267. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Staudt, T.; Engler, A.; Rittweger, E.; Harke, B.; Engelhardt, J.; Hell, S.W. Far-field optical nanoscopy with reduced number of state transition cycles (REScue). Opt. Express 2011, 19, 5644–5657. [Google Scholar] [CrossRef] [PubMed]
- Heine, J.; Reuss, M.; Harke, B.; D’Este, E.; Sahl, S.J.; Hell, S.W. Adaptive illumination STED nanoscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 9797–9802. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers1, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 345, 606–612. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef]
- Cremer, C.; Masters, B.R. Resolution enhancement techniques in microscopy. Eur. Phys. J. H 2013, 38, 281–344. [Google Scholar] [CrossRef]
- Müller, C.B.; Enderlein, J. Image scanning microscopy. Phys. Rev. Lett. 2010, 104, 198101. [Google Scholar] [CrossRef]
- de Luca, G.M.R.; Breedijk, R.M.P.; Brandt, R.A.J.; Zeelenberg, C.H.C.; de Jong, B.E.; Timmermans, W.; Nahidi Azar, L.; Hoebe, R.A.; Stallinga, S.; Manders, E.M.M. Re-scan confocal microscopy: Scanning twice for better resolution. Biomed. Opt. Express 2013, 4, 2644–2656. [Google Scholar] [CrossRef]
- Heintzmann, R.; Cremer, C. Laterally modulated excitation microscopy: Improvement of resolution by using a diffraction grating. In Optical Biopsies and Microscopic Techniques III, Proceedings of the SPIE 3568, Stockholm, Sweden, 8–12 September 1998; SPIE: Bellingham, WA, USA, 1999; pp. 185–196. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Lu-Walther, H.-W.; Jost, A.; Kielhorn, M.; Wicker, K.; Heintzmann, R. Simple structured illumination microscope setup with high acquisition speed by using a spatial light modulator. Opt. Express 2014, 22, 20663–20677. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, R.; Liu, W.; Zhu, D.; Zhang, Z.; Kuang, C.; Liu, X. Widefield and total internal reflection fluorescent structured illumination microscopy with scanning galvo mirrors. J. Biomed. Opt. 2018, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schneckenburger, H.; Richter, V.; Wagner, M. Live-Cell Optical Microscopy with Limited Light Doses; SPIE Spotlight Series; SPIE: Bellingham, WA, USA, 2018; Volume SL 42. [Google Scholar] [CrossRef]
- Demmerle, J.; Innocent, C.; North, A.J.; Ball, G.; Müller, M.; Miron, E.; Matsuda, A.; Dobbie, I.M.; Markaki, Y.; Schermelleh, L. Strategic and practical guidelines for successful structured illumination microscopy. Nat. Protoc. 2017, 12, 988–1010. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Mönkemöller, V.; Hennig, S.; Hübner, W.; Huser, T. Open-source image reconstruction of super-resolution structured illumination microscopy data in ImageJ. Nat. Commun. 2016, 7, 10980. [Google Scholar] [CrossRef] [PubMed]
- Komis, G.; Mistrik, M.; Šamajová, O.; Doskocilová, A.; Ovecka, M.; Illés, P.; Bartek, J.; Šamaj, J. Dynamics and organization of cortical microtubules as revealed by superresolution structured illumination Microscopy. Plant Physiol. 2014, 165, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 2018, 175, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.H.; Carter, S.K. Adriamycin. A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1974, 80, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Wang, T.T.; Wu, Y.T.; Xu, C.M.; Dong, M.Y.; Sheng, J.Z.; Huang, H.F. Adriamycin induces H2AX phosphorylation in human spermatozoa. Asian J. Androl. 2008, 10, 749–757. [Google Scholar] [CrossRef]
- Scaduto, R.C., Jr.; Grotyohann, L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Gschwend, M.H.; Strauss, W.S.L.; Sailer, R.; Kron, M.; Steeb, U.; Steiner, R. Energy transfer spectroscopy for measuring mitochondrial metabolism in living cells. Photochem. Photobiol. 1997, 66, 34–41. [Google Scholar] [CrossRef]
- Pinotsi, D.; Buell, A.K.; Galvagnion, C.; Dobson, C.M.; Gabriele, S.; Kaminski-Schierle, G.S.; Kaminski, C.F. Direct Observation of Heterogeneous Amyloid Fibril Growth Kinetics via Two-Color Super-Resolution Microscopy. Nano Lett. 2014, 14, 339–345. [Google Scholar] [CrossRef]
- Heintzmann, R.; Huser, T. Super-resolution structured illumination microscopy. Chem. Rev. 2017, 117, 13890–13908. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; So, P.T.C. Three-dimensional super-resolution high-throughput imaging by structured illumination STED microscopy. Opt. Express 2018, 26, 20920–20928. [Google Scholar] [CrossRef] [PubMed]
- Lu-Walther, H.-W.; Kielhorn, M.; Förster, R.; Jost, A.; Wicker, K.; Heintzmann, R. fastSIM: A practical implementation of fast structured illumination microscopy. Methods Appl. Fluoresc. 2015, 3, 014001. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E. Excitation strategies for optical lattice microscopy. Opt. Express 2005, 13, 3021–3036. [Google Scholar] [CrossRef] [PubMed]
- Godin, A.G.; Lounis, B.; Cognet, L. Super-resolution Microscopy Approaches for Live Cell Imaging. Biophys. J. 2014, 107, 1777–1784. [Google Scholar] [CrossRef]
- Tønnesen, J.; Krishna Inavalli, V.V.G.; Nägerl, U.V. Super-Resolution Imaging of the Extracellular Space in Living Brain Tissue. Cell 2018, 172, 1108–1121. [Google Scholar] [CrossRef] [PubMed]
- Young, L.J.; Ströhl, F.; Kaminski, C.F. A guide to structured illumination TIRF microscopy at high speed with multiple colors. J. Vis. Exp. 2016, 30, 53988. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, V.; Piper, M.; Wagner, M.; Schneckenburger, H. Increasing Resolution in Live Cell Microscopy by Structured Illumination (SIM). Appl. Sci. 2019, 9, 1188. https://doi.org/10.3390/app9061188
Richter V, Piper M, Wagner M, Schneckenburger H. Increasing Resolution in Live Cell Microscopy by Structured Illumination (SIM). Applied Sciences. 2019; 9(6):1188. https://doi.org/10.3390/app9061188
Chicago/Turabian StyleRichter, Verena, Mathis Piper, Michael Wagner, and Herbert Schneckenburger. 2019. "Increasing Resolution in Live Cell Microscopy by Structured Illumination (SIM)" Applied Sciences 9, no. 6: 1188. https://doi.org/10.3390/app9061188
APA StyleRichter, V., Piper, M., Wagner, M., & Schneckenburger, H. (2019). Increasing Resolution in Live Cell Microscopy by Structured Illumination (SIM). Applied Sciences, 9(6), 1188. https://doi.org/10.3390/app9061188