
New Steroid–Alkaloid Bioconjugates as Potential Bioactive Compounds: Synthesis, Spectroscopic and In Silico Study

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
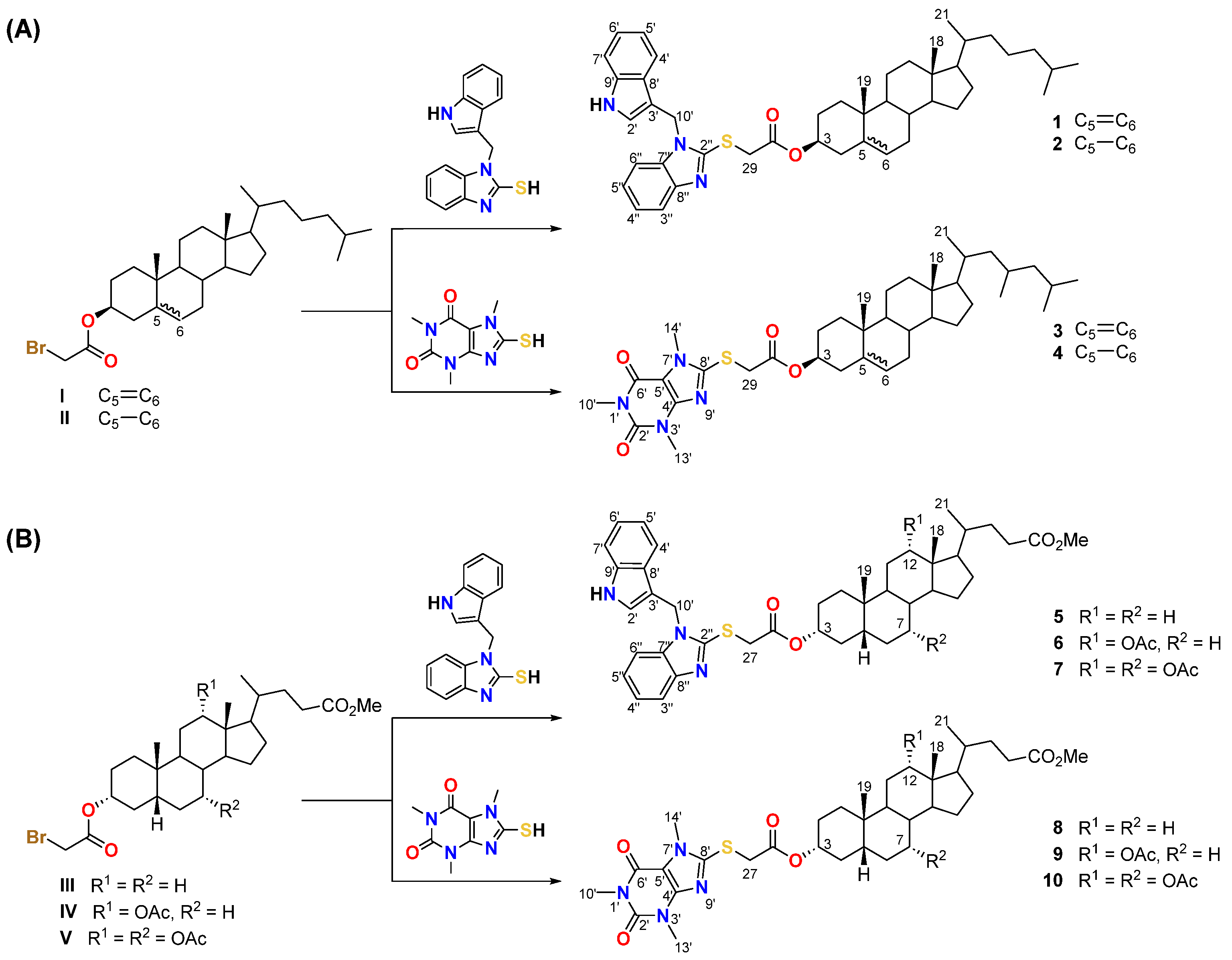
2.2. Synthesis
2.3. PM5 Calculation
2.4. PASS Analysis
2.5. Docking Study
3. Results and Discussion
3.1. Synthesis and Spectroscopic Characterization
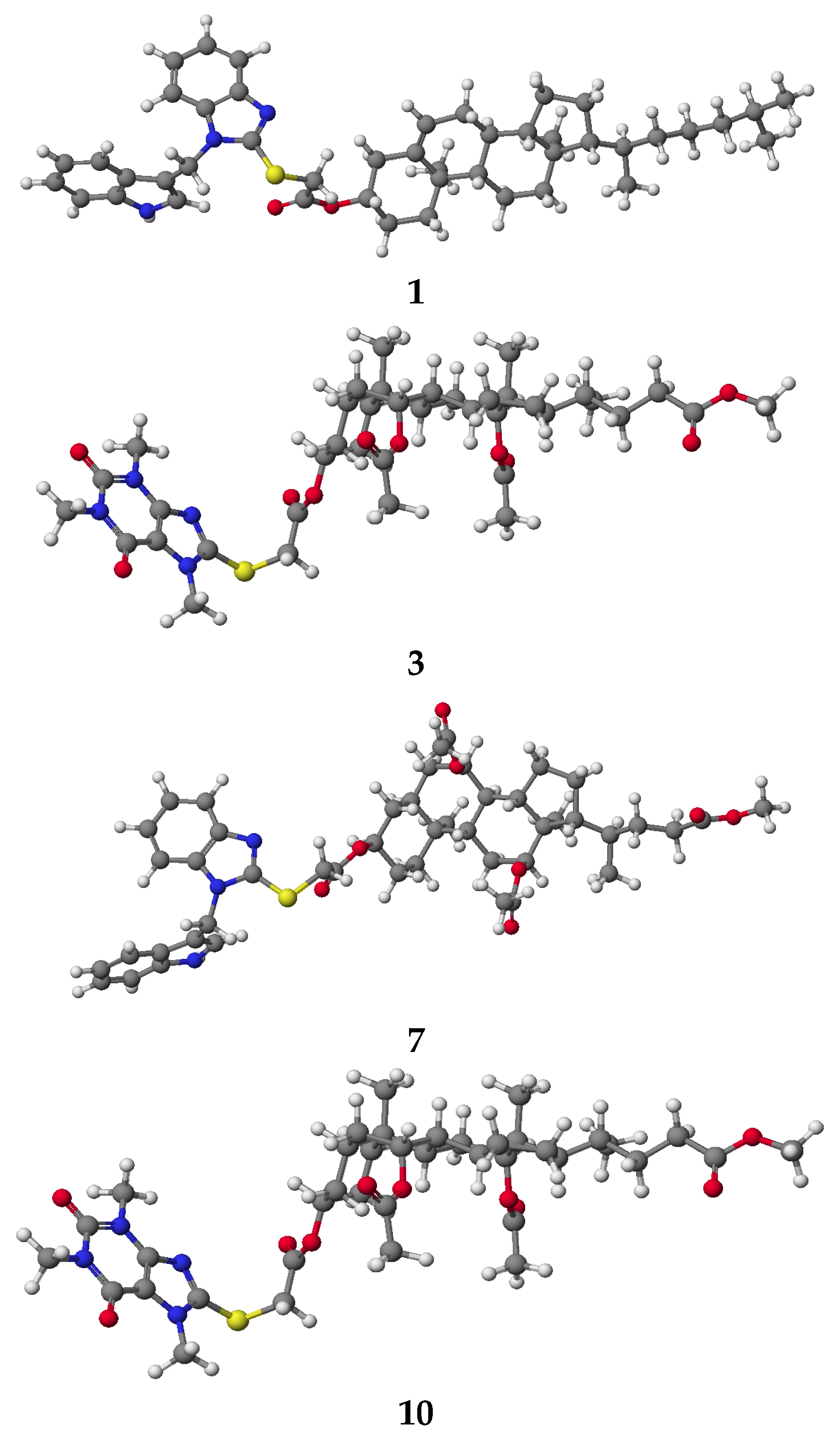
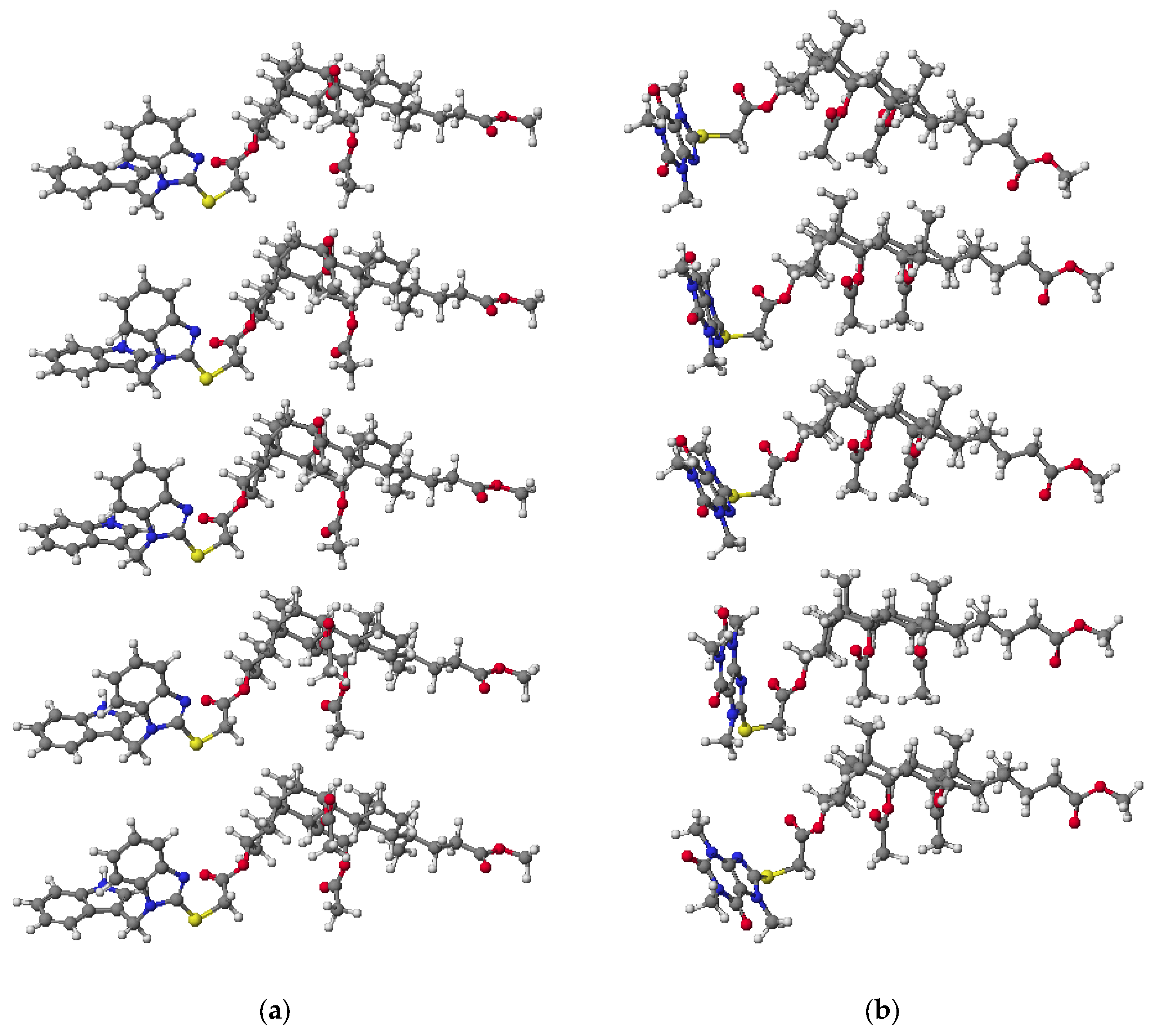
3.2. PM5 Calculation
3.3. PASS Analysis
3.4. Docking Study
3.4.1. Selected Bioconjugates and Protein Domains
- −
- HMG-CoA reductase activity (PDB ID: 1HWK): This enzyme plays a pivotal role in the mevalonate pathway, catalyzing HMG-CoA conversion to mevalonate, a key precursor in cholesterol biosynthesis. The structure of HMG-CoA reductase in complex with statins reveals their mechanism of inhibition and provides valuable insights into the design of drugs for treating hypercholesterolemia and cardiovascular disease. Understanding the binding interactions at the active site can lead to developing more effective and selective inhibitors that minimize the side effects associated with cholesterol-lowering therapies [44,45].
- −
- Cysteinyl leukotriene receptor 1 (CysLT1R) activity (PDB ID: 6RZ4): This G protein-coupled receptor is crucial for mediating inflammatory responses, particularly in asthma and allergic reactions. The crystal structure of CysLT1R complexed with the antagonists zafirlukast and pranlukast elucidates the binding mechanisms and conformational changes associated with receptor inhibition. These insights are essential for the design of new therapeutic agents targeting CysLT1R, potentially leading to more effective treatments for asthma and other inflammatory diseases [46,47].
3.4.2. Similarities and Differences Between Novel and Endogenous Ligands
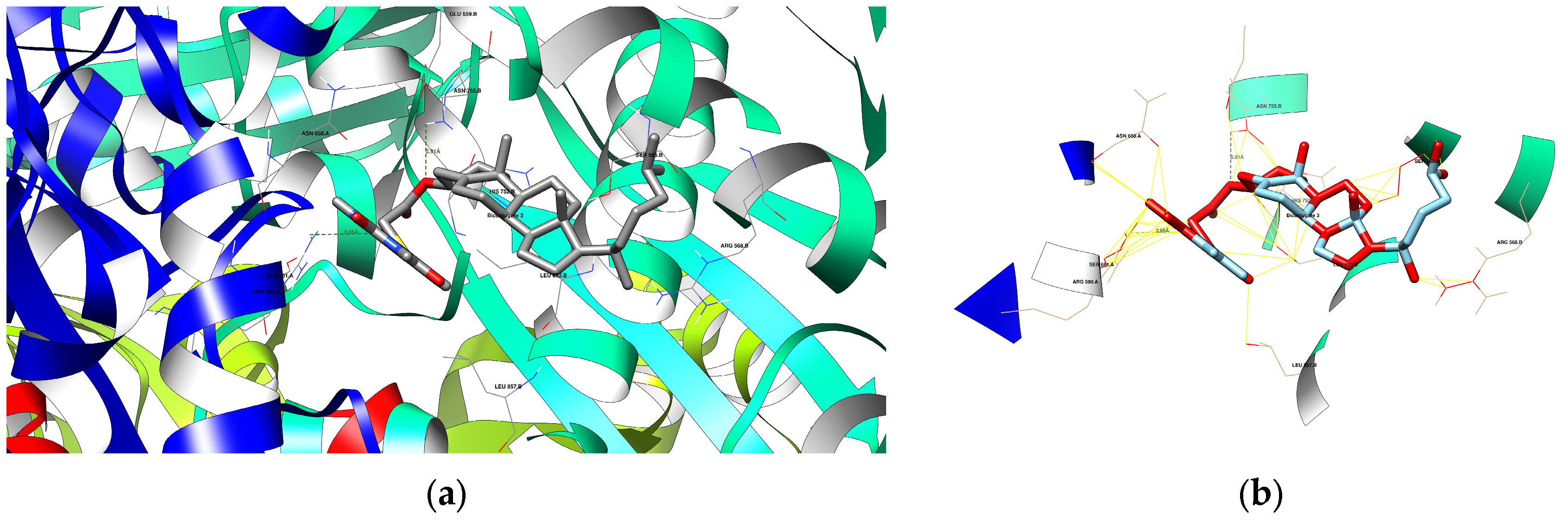
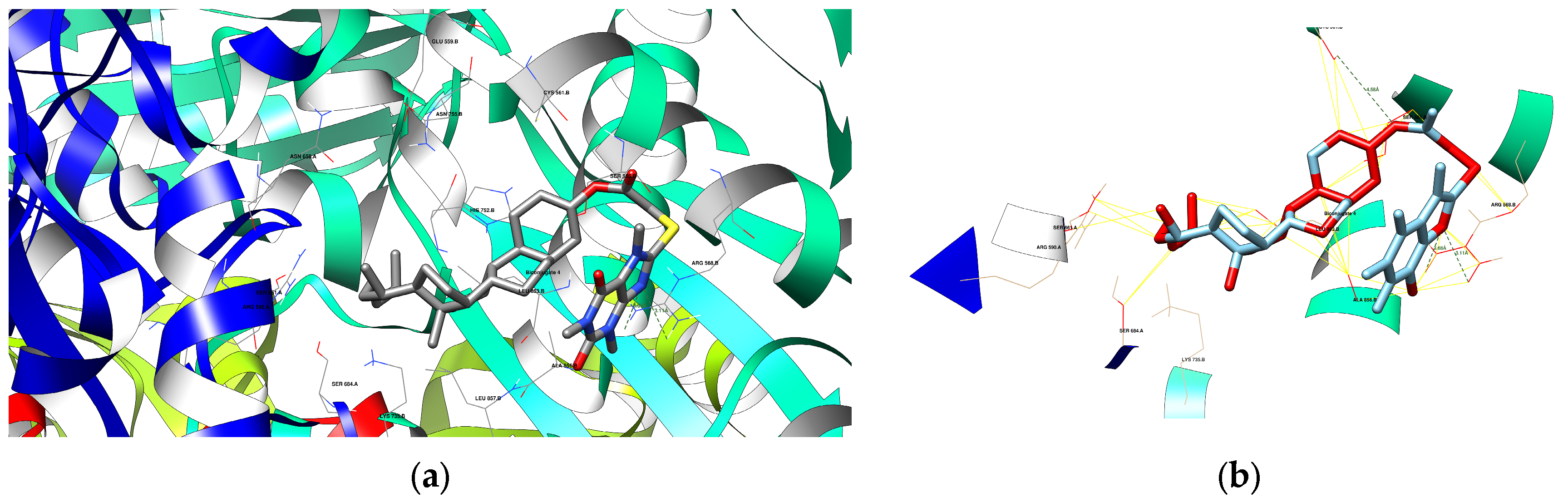
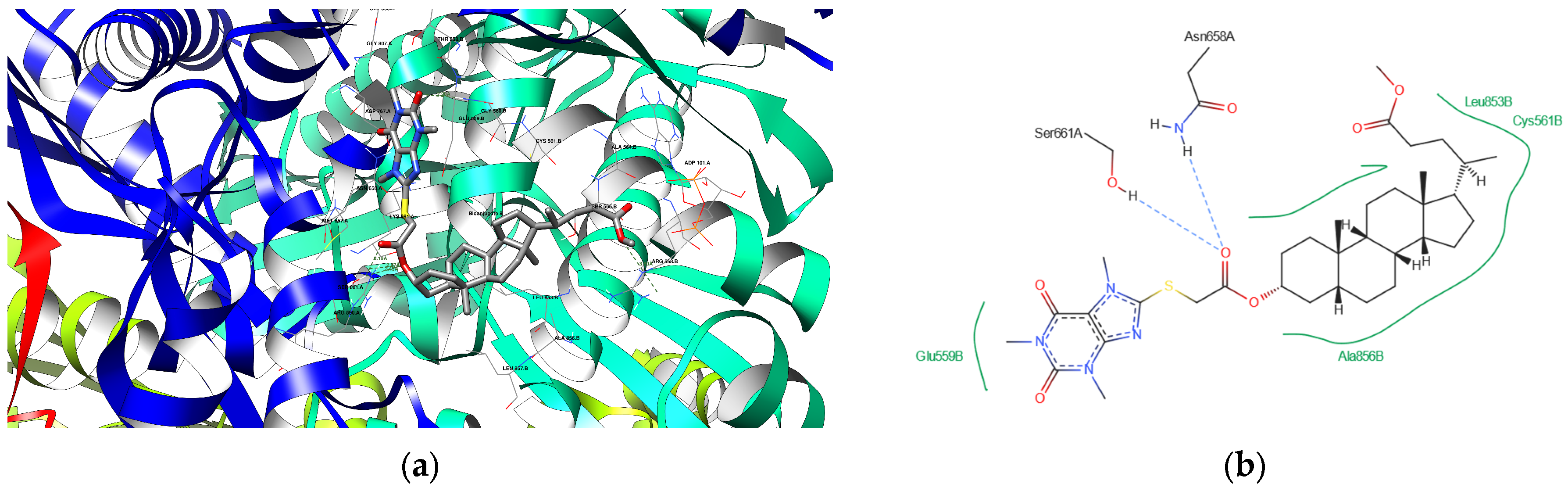
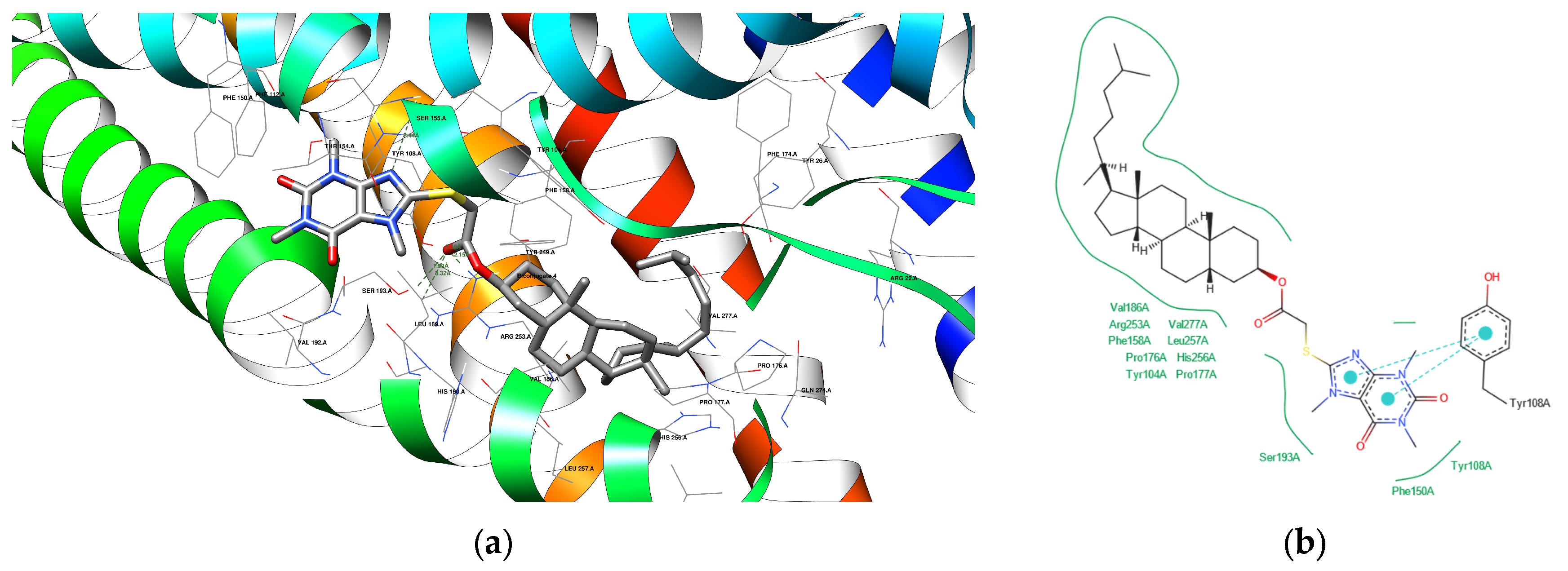
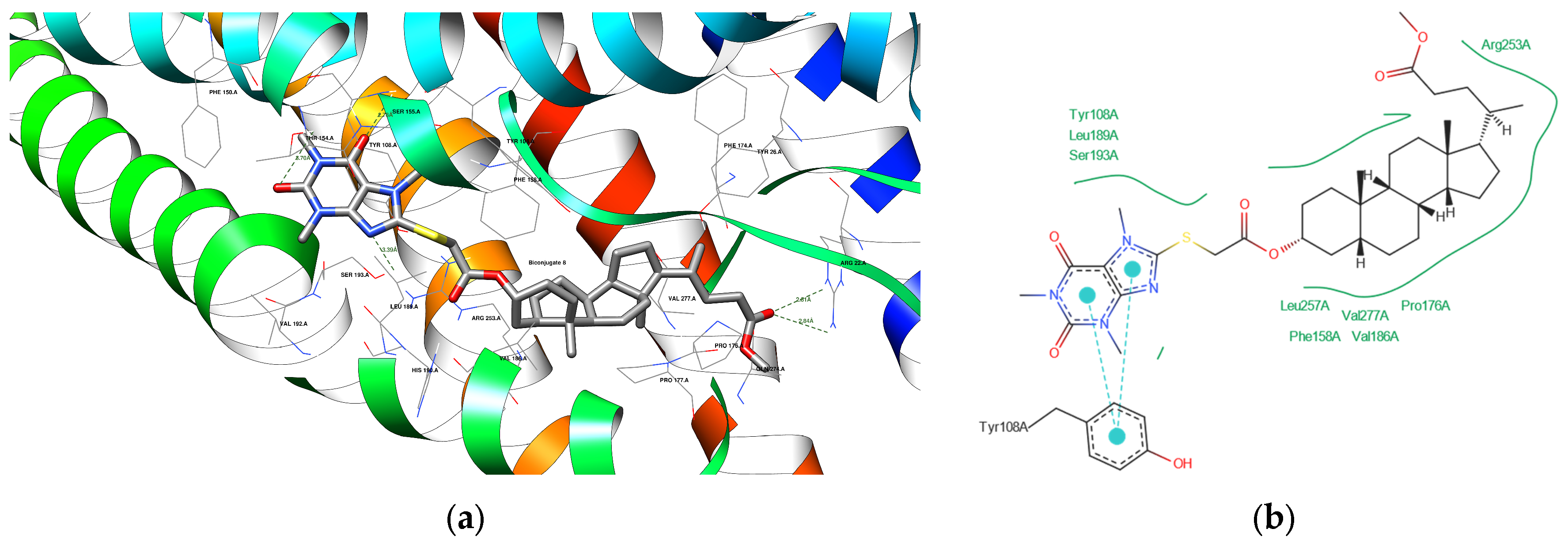
3.4.3. Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lednicer, D. Strategies for Organic Drug Synthesis and Design; John Wiley & Sons Ltd.: Chichester, UK, 2011. [Google Scholar]
- Santiago-Sampedro, G.I.; Aguilar-Granda, A.; Torres-Huerta, A.; Flores-Álamo, M.; Maldonado-Domínguez, M.; Rodríguez-Molina, B.; Iglesias-Arteaga, A. Self-assembly of an amphiphilic bile acid dimer: A combined experimental and theoretical study of its medium-responsive fluorescence. J. Org. Chem. 2022, 87, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Bariya, D.; Mishra, R.; Mishra, B. Bile acid-based receptors and their applications in recognition. Steroids 2022, 179, 108981. [Google Scholar] [CrossRef]
- Jain, M.; Nowak, B.P.; Ravoo, B.J. Supramolecular hydrogels based on cyclodextrins: Progress and perspectives. Chem. Nano Mat. 2022, 8, e202200077. [Google Scholar] [CrossRef]
- Kovacevic, B.; Jones, M.; Ionsecu, C.; Walker, D.; Wagle, S.; Chester, J.; Foster, T.; Brown, D.; Mikov, M.; Mooranian, A.; et al. The effect of deoxycholic acid on chitosan-enabled matrices for tissue scaffolding and injectable nanogels. Biomaterials 2022, 8, 358. [Google Scholar] [CrossRef]
- Gao, H.; Dias, J.R. Synthesis and characterization of dimeric bile acid ester derivatives. J. Für Prakt. Chem. Chem.-Ztg. 1997, 339, 187–190. [Google Scholar] [CrossRef]
- Li, Y.; Dias, J.R. Syntheses of α-and β-dimers of lithocholic acid esters. Org. Prep. Proced. Int. 1996, 28, 203–209. [Google Scholar] [CrossRef]
- Paryzek, Z.; Joachimiak, R.; Piasecka, M.; Pospieszny, T. A new approach to steroid dimers and macrocycles by the reaction of 3-chlorocarbonyl derivatives of bile acids with O,O-, N,N-, and S,S-dinucleophiles. Tetrahedron Lett. 2012, 53, 6212–6215. [Google Scholar] [CrossRef]
- Wallimann, P.; Marti, T.; Fürer, A.; Diederich, F. Steroids in molecular recognition. Chem. Rev. 1997, 97, 1567–1608. [Google Scholar] [CrossRef]
- Tamminen, J.; Kolehmainen, E. Bile acids as building blocks of supramolecular hosts. Molecules 2001, 6, 21–46. [Google Scholar] [CrossRef]
- Nagrady, T.; Weaver, D.F. Medicinal Chemistry: A Molecular and Biochemical Approach, 3rd ed.; Oxford University Press: New York, NY, USA, 2005; pp. 316–320. [Google Scholar]
- Parish, E.J.; Nes, W.D. Biochemistry and Function of Sterols; CRC Press: Boca Raton, FL, USA, 1997. [Google Scholar]
- Schaller, H. The role of sterols in plant growth and development. Prog. Lipid Res. 2003, 42, 163–175. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Elshemy, H.A.H.; El-Nahass, E.S.; Abdel-Fattah, M.M.; Abdelgawad, Y.Y.M. New indomethacin analogs as selective COX-2 inhibitors: Synthesis, COX-1/2 inhibitory activity, anti-inflammatory, ulcerogenicity, histopathological, and docking studies. Arch. Der Pharm. 2021, 354, 2000328. [Google Scholar] [CrossRef]
- Jagadeesan, S.; Karpagam, S. Novel series of N-acyl substituted indole-based piperazine, thiazole, and tetrazoles as potential antibacterial, antifungal, antioxidant, and cytotoxic agents, and their docking investigation as potential Mcl-1 inhibitors. J. Mol. Struct. 2023, 1271, 134013. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Laubach, J.P.; Richter, J.; Stricker, S.; Spencer, A.; Richardson, P.G.; Chari, A. Panobinostat from bench to bedside: Rethinking the treatment paradigm for multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 752–765. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, Q.; Li, N.; Gu, L.; Dan, W.; Dai, J. Recent developments of gramine: Chemistry and biological activity. Molecules 2023, 28, 5695. [Google Scholar] [CrossRef] [PubMed]
- Centofanti, F.; Alonzi, T.; Latini, A.; Spitalieri, P.; Murdocca, M.; Chen, X.; Cui, W.; Shang, Q.; Goletti, D.; Shi, Y.; et al. Indole-3-carbinol In Vitro antiviral activity against SARS-CoV-2 virus and in vivo toxicity. Cell Death Discov. 2022, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.R.; Tsai, C.H.; Kulp, S.K.; Chen, C.S. Indole-3-carbinol as a chemopreventive and anti-cancer agent. Cancer Lett. 2008, 262, 153–163. [Google Scholar] [CrossRef]
- Cai, Q.N.; Han, Y.; Cao, Y.Z.; Hu, Y.; Zhao, X.; Bi, J.L. Detoxification of gramine by the cereal aphid Sitobion avenae. J. Chem. Ecol. 2009, 35, 320–325. [Google Scholar] [CrossRef]
- Quartarone, G.; Ronchin, L.; Vavasori, A.; Tortato, C.; Bonaldo, L. Inhibitive action of gramine towards corrosion of mild steel in deaerated 1.0 M hydrochloric acid solutions. Corros. Sci. 2012, 64, 82–89. [Google Scholar] [CrossRef]
- Kozanecka-Okupnik, W.; Jasiewicz, B.; Pospieszny, T.; Matuszak, M.; Mrówczyńska, L. Haemolytic activity of formyl- and acetyl-derivatives of bile acids and their gramine salts. Steroids 2017, 126, 50–56. [Google Scholar] [CrossRef]
- Kozanecka, W.; Mrówczyńska, L.; Pospieszny, T.; Jasiewicz, B.; Gierszewski, M. Synthesis, spectroscopy, theoretical and biological studies of new gramine-steroids salts and conjugates. Steroids 2015, 98, 92–99. [Google Scholar] [CrossRef]
- Fiani, B.; Zhu, L.; Musch, B.L.; Briceno, S.; Andel, R.; Sadeq, N.; Ansari, A.Z. The neurophysiology of caffeine as a central nervous system stimulant and the resultant effects on cognitive function. Cureus 2021, 13, e15032. [Google Scholar] [CrossRef] [PubMed]
- Woziwodzka, A.; Krychowiak-Maśnicka, M.; Gołuński, G.; Łosiewska, A.; Borowik, A.; Wyrzykowski, D.; Piosik, J. New life of an old drug: Caffeine as a modulator of antibacterial activity of commonly used antibiotics. Pharmaceuticals 2022, 15, 872. [Google Scholar] [CrossRef] [PubMed]
- AlEraky, D.M.; Abuohashish, H.M.; Gad, M.M.; Alshuyukh, M.H.; Bugshan, A.S.; Almulhim, K.S.; Mahmoud, M.M. The antifungal and antibiofilm activities of caffeine against Candida albicans on polymethyl methacrylate denture base material. Biomedicines 2022, 10, 2078. [Google Scholar] [CrossRef]
- Sierakowska, A.; Jasiewicz, B.; Piosik, Ł.; Mrówczyńska, L. New C8-substituted caffeine derivatives as promising antioxidants and cytoprotective agents in human erythrocytes. Sci. Rep. 2023, 13, 1785. [Google Scholar] [CrossRef]
- Jasiewicz, B.; Sierakowska, A.; Wandyszewska, N.; Warżajtis, B.; Rychlewska, U.; Wawrzyniak, R.; Mrówczyńska, L. Antioxidant properties of thio-caffeine derivatives: Identification of the newly synthesized 8-[(pyrrolidin-1-ylcarbonothioyl)sulfanyl] caffeine as antioxidant and highly potent cytoprotective agent. Bioorg. Med. Chem. Lett. 2016, 26, 3994–3998. [Google Scholar] [CrossRef]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xy, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N.; Schwarzschild, M.A. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 15, RC143. [Google Scholar] [CrossRef]
- Booysen, H.P.; Moraal, C.; Terre’Blanche, G.; Petzer, A.; Bergh, J.J.; Petzer, J.P. Thio- and aminocaffeine analogues as inhibitors of human monoamine oxidase. Bioorg. Med. Chem. 2011, 19, 7507–7518. [Google Scholar] [CrossRef]
- Soltani Rad, M.N.; Behrouz, S.; Aghajani, S.; Behrouz, M.; Zarenezhad, E.; Ghanbariasad, A. Design, synthesis, anticancer and in silico assessment of 8-caffeinyl-triazolylmethoxy hybrid conjugates. RSC Adv. 2023, 13, 3056–3070. [Google Scholar] [CrossRef]
- Reshetnikov, D.V.; Burova, L.G.; Rybalova, T.V.; Bondareva, E.A.; Patrushev, S.S.; Evstropov, A.N.; Shults, E.E. Synthesis and Antibacterial Activity of Caffeine Derivatives Containing Amino-Acid Fragments. Chem. Nat. Compd. 2022, 58, 800–806. [Google Scholar] [CrossRef]
- Ohshita, K.; Ishiyama, H.; Oyanagi, K.; Nakata, H.; Kobayashi, J. Synthesis of hybrid molecules of caffeine and eudistomin D and its effects on adenosine receptors. Bioorg. Med. Chem. 2007, 15, 3235–3240. [Google Scholar] [CrossRef]
- Kozanecka-Okupnik, W.; Jasiewicz, B.; Pospieszny, T.; Jastrząb, R.; Skrobańska, M.; Mrówczyńska, L. Spectroscopy, molecular modeling, and antioxidant activity studies on novel conjugates containing indole and uracil moiety. J. Mol. Struct. 2018, 1169, 130–137. [Google Scholar] [CrossRef]
- Jasiewicz, B.; Babijczuk, K.; Warżajtis, B.; Rychlewska, U.; Starzyk, J.; Cofta, G.; Mrówczyńska, L. Indole derivatives bearing imidazole, benzothiazole-2-thione, or benzoxazole-2-thione moieties—Synthesis, structure, and evaluation of their cytoprotective, antioxidant, antibacterial, and fungicidal activities. Molecules 2023, 28, 708. [Google Scholar] [CrossRef]
- Berdzik, N.; Koenig, H.; Mrowczyńska, L.; Nowak, D.; Jasiewicz, B.; Pospieszny, T. Synthesis and hemolytic activity of bile acid-indole bioconjugates linked by triazole. J. Org. Chem. 2023, 88, 16719–16734. [Google Scholar] [CrossRef] [PubMed]
- Babijczuk, K.; Berdzik, N.; Nowak, D.; Warżajtis, B.; Rychlewska, U.; Starzyk, J.; Mrówczyńska, L.; Jasiewicz, B. Novel C3-methylene-bridged indole derivatives with and without substituents at N1: The influence of substituents on their hemolytic, cytoprotective, and antimicrobial activity. Int. J. Mol. Sci. 2024, 25, 5364. [Google Scholar] [CrossRef]
- Yan, B.; Liu, L.; Huang, S.; Ren, Y.; Wang, H.; Yao, Z.; Li, L.; Chen, S.; Wang, X.; Zhang, Z. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 2017, 53, 3637. [Google Scholar] [CrossRef]
- Kawka, A.; Hajdaś, G.; Kułaga, D.; Koenig, H.; Kowalczyk, I.; Pospieszny, T. Molecular structure, spectral and theoretical study of new type bile acid–sterol conjugates linked via 1,2,3-triazole ring. J. Mol. Struct. 2023, 1273, 134313. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 5. [Google Scholar] [CrossRef]
- Raschka, S. Biopandas: Working with molecular structures in pandas dataframes. J. Open Source Softw. 2017, 2, 14. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Complex of the catalytic portion of human HMG-CoA reductase with atorvastatin. Protein Data Bank. 2001. Available online: https://www.wwpdb.org/pdb?id=pdb_00001hwk (accessed on 3 December 2024). [CrossRef]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Besserer-Offroy, É.; Longpré, J.-M.; Lyapina, E.; et al. Structure-Based Mechanism of Cysteinyl Leukotriene Receptor Inhibition by Antiasthmatic Drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiryaeva, A.; Besserer-Offroy, É.; Longpré, J.-M.; Lyapina, E.; et al. Crystal Structure of Cysteinyl Leukotriene Receptor 1 in Complex with Pranlukast. Protein Data Bank. 2019. Available online: https://www.wwpdb.org/pdb?id=pdb_00006rz4 (accessed on 3 December 2024).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDock-Tools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- ProteinsPlus Development Team. ProteinsPlus. Available online: https://proteins.plus (accessed on 18 November 2024).
- Schöning-Stierand, K.; Diedrich, K.; Ehrt, C.; Flachsenberg, F.; Graef, J.; Sieg, J.; Penner, P.; Poppinga, M.; Ungethüm, A.; Rarey, M. ProteinsPlus: A comprehensive collection of web-based molecular modeling tools. Nucleic Acids Res. 2022, 50, W611–W615. [Google Scholar] [CrossRef]
- Stierand, K.; Maaß, P.C.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef]
- Diedrich, K.; Krause, B.; Berg, O.; Rarey, M. PoseEdit: Enhanced ligand binding mode communication by interactive 2D diagrams. J. Comput. Aided Mol. Des. 2023, 37, 491–503. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A. Predictive Toxicology; Helma, C., Ed.; Taylor & Francis: Boca Raton, FL, USA, 2005; pp. 459–478. [Google Scholar]
- Poroikov, V.V.; Filimonov, D.A. How to acquire new biological activities in old compounds by computer prediction. J. Comput. Aided Mol. Des. 2002, 16, 819–824. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Borodina, Y.V.; Lagunin, A.A.; Kos, A. Robustness of biological activity spectra predicting by computer program PASS for non-congeneric sets of chemical compounds. J. Chem. Inf. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef]
- Mohammed, A.I.; Musa, A.; Abu-Bakr, M.S.; Abbass, H.S. Anti-eczematic and molecular modeling of anthraquinones isolated from the seeds of Asphodelus microcarpus salzm. viv. growing in Egypt. Pharmacogn. Mag. 2019, 15, 586–591. [Google Scholar] [CrossRef]
- Watschinger, K.; Keller, M.A.; Hermetter, A.; Golderer, G.; Werner-Felmayer, G.; Werner, E.R. Glyceryl ether monooxygenase resembles aromatic amino acid hydroxylases in metal ion and tetrahydrobiopterin dependence. Biol. Chem. 2009, 390, 3–10. [Google Scholar] [CrossRef]
- Rahmati-Ahmadabad, S.; Broom, D.R.; Ghanbari-Niaki, A.; Shirvani, H. Effects of exercise on reverse cholesterol transport: A systemized narrative review of animal studies. Life Sci. 2019, 224, 139–148. [Google Scholar] [CrossRef]
- Agosto, N.J.; Alambatin, P.B.; Bacalso, J.; Cabisada, J.; Carating, B.D. The evaluation of Citrus bergamia phytochemicals as potential cholesterol-lowering agents against HMG-CoA reductase: An in silico molecular docking study. Biol. Life Sci. Forum 2024, 35, 7. [Google Scholar] [CrossRef]
- Peppin, J.F.; Pergolizzi, J.V.; Fudin, J.; Meyer, T.A.; Raffa, R.B. History of respiratory stimulants. J. Pain Res. 2021, 14, 1043–1049. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics Software; Zenodo: Geneva, Switzerland, 2022. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: An analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen bonds: Simple after all? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Search Space Center (x, y, z) | Size of the Search Space (x, y, z) |
|---|---|---|
| 1HWK | 18, 8, 15 | 42, 41, 39 |
| 6RZ4 | 40, 28, 57 | 36, 44, 45 |
| Compound | HOF [kcal/mol] |
|---|---|
| 1 | −67.9848 |
| 2 | −90.5785 |
| 3 | −205.2146 |
| 4 | −227.2060 |
| 5 | −156.9531 |
| 6 | −242.8710 |
| 7 | −327.7994 |
| 8 | −293.8754 |
| 9 | −378.4569 |
| 10 | –462.4638 |
| Pa > 70% | Bioconjugates | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Cholesterol antagonist | 82 | 76 | 95 | 94 | 74 | – | – | 94 | 92 | 91 |
| Analeptic | – | – | 73 | 79 | – | – | – | 82 | 83 | 75 |
| Antihypercholesterolemic | – | – | 91 | 84 | – | – | – | 78 | – | 72 |
| Respiratory analeptic | – | – | 85 | 88 | – | – | – | 87 | 82 | 70 |
| Lipoprotein lipase inhibitor | – | – | – | 84 | – | – | – | 78 | 75 | – |
| Acylcarnitine hydrolase Inhibitor | – | – | – | 71 | – | – | – | 75 | 72 | 75 |
| Glyceryl-ether monooxygenase inhibitor | – | – | – | – | – | – | 73 | – | 71 | 74 |
| Hypolipemic | – | – | – | – | – | – | – | – | – | 74 |
| CYP3A1 substrate | – | – | 75 | – | – | – | – | – | – | |
| CYP3A4 inducer | – | – | 70 | – | – | – | – | – | – | – |
| CYP3A inducer | – | – | 70 | – | – | – | – | – | – | – |
| Alkylacetylglycerophosphatase Inhibitor | – | – | – | 72 | – | – | – | – | – | – |
| Cytoprotectant | – | – | – | – | 70 | – | 70 | – | – | – |
| Antieczematic | – | – | – | – | 71 | – | – | – | – | – |
| Compound | Molecular Weight [g/mol] | HB Acceptors | HB Donors | Polar Surface Area [Å2] | aLogP | Rotatable Bonds |
|---|---|---|---|---|---|---|
| Atorvastatin | 559 | 5 | 4 | 111.79 | 6.31 | 12 |
| 1 | 706 | 4 | 1 | 59.91 | 11.61 | 11 |
| 3 | 653 | 6 | 0 | 88.12 | 7.02 | 9 |
| 4 | 655 | 6 | 0 | 88.12 | 7.10 | 9 |
| 8 | 657 | 8 | 0 | 114.42 | 5.22 | 8 |
| Caffeine | 194 | 3 | 0 | 61.82 | −1.03 | 1 |
| PDB ID | Compound | Binding Energy [kcal/mol] | Standard Deviation of Binding Energy [kcal/mol] |
|---|---|---|---|
| 1HWK | Atorvastatin | −9.6 | 0.56 |
| 1 | −8.4 | 0.17 | |
| 3 | −8.0 | 0.12 | |
| 4 | −9.0 | 0.23 | |
| 8 | −8.7 | 0.35 | |
| Caffeine | −6.1 | 0.19 | |
| 6RZ4 | Caffeine | −6.2 | 0.21 |
| 4 | −9.9 | 0.78 | |
| 8 | −11.2 | 0.97 | |
| Pranlukast | −12.7 | 0.98 |
| Compound | Protein Domain | Amino Acid Residues | Distance [Å] | Is Compound Donor or Acceptor? |
|---|---|---|---|---|
| 1 | 1HWK | ARG 568 B SER 565 B | 3.33 2.73 | Acceptor Donor |
| 3 | 1HWK | ASN 755 B ARG 590 A | 2.91 2.85 | Acceptor Acceptor |
| 4 | 1HWK | ARG 568 B | 3.11 | Acceptor |
| 8 | 1HWK | GLY 560 B ASN 658 A SER 661 A ARG 590 A | 2.20 3.48 2.19 2.47 | Acceptor Acceptor Acceptor Acceptor |
| 4 | 6RZ4 | SER 155 A SER 193 A | 3.44 1.99 | Acceptor Acceptor |
| 8 | 6RZ4 | THR 154 A SER 155 A SER 193 A ARG 22 A | 3.70 2.78 3.39 2.81 | Acceptor Acceptor Acceptor Acceptor |
| Atorvastatin | 1HWK | SER 565 B ASN 755 B ASP 690 A SER 684 A LYS 735 B | 2.21 2.05 2.36 2.09 1.91 | Acceptor Acceptor Donor Acceptor Acceptor |
| Caffeine | 6RZ4 | SER 155 A | 2.80 | Acceptor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koenig, H.; Babijczuk, K.; Ostrowski, K.; Nowak, D.; Pospieszny, T.; Jasiewicz, B. New Steroid–Alkaloid Bioconjugates as Potential Bioactive Compounds: Synthesis, Spectroscopic and In Silico Study. Appl. Sci. 2025, 15, 591. https://doi.org/10.3390/app15020591
Koenig H, Babijczuk K, Ostrowski K, Nowak D, Pospieszny T, Jasiewicz B. New Steroid–Alkaloid Bioconjugates as Potential Bioactive Compounds: Synthesis, Spectroscopic and In Silico Study. Applied Sciences. 2025; 15(2):591. https://doi.org/10.3390/app15020591
Chicago/Turabian StyleKoenig, Hanna, Karolina Babijczuk, Kamil Ostrowski, Damian Nowak, Tomasz Pospieszny, and Beata Jasiewicz. 2025. "New Steroid–Alkaloid Bioconjugates as Potential Bioactive Compounds: Synthesis, Spectroscopic and In Silico Study" Applied Sciences 15, no. 2: 591. https://doi.org/10.3390/app15020591
APA StyleKoenig, H., Babijczuk, K., Ostrowski, K., Nowak, D., Pospieszny, T., & Jasiewicz, B. (2025). New Steroid–Alkaloid Bioconjugates as Potential Bioactive Compounds: Synthesis, Spectroscopic and In Silico Study. Applied Sciences, 15(2), 591. https://doi.org/10.3390/app15020591