
From Dysbiosis to Cardiovascular Disease: The Impact of Gut Microbiota on Atherosclerosis and Emerging Therapies

 ,
,  , ,
, ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Overview of the Human Gut Microbiota
2.1. Development and Maturation of the Gut Microbiota
2.2. Composition and Functional Organisation of the Human Gut Microbiota
2.2.1. Metabolic Functions
2.2.2. Regulation of Immune Response and Protection Against Pathogenic Organisms
2.3. Factors Influencing the Gut Microbiota
2.3.1. Exposure to Antibiotics
2.3.2. Diet
2.3.3. Other Factors
3. Cardiovascular Diseases
3.1. Atherosclerosis
3.1.1. Endothelium
3.1.2. Endothelial Dysfunction
3.1.3. Formation of Fatty Streaks
3.1.4. Lesion Progression
3.1.5. Plaque Rupture and Thrombus Formation
4. Interaction Between the Gut Microbiota and Cardiovascular Diseases
4.1. Trimethylamine N-Oxide
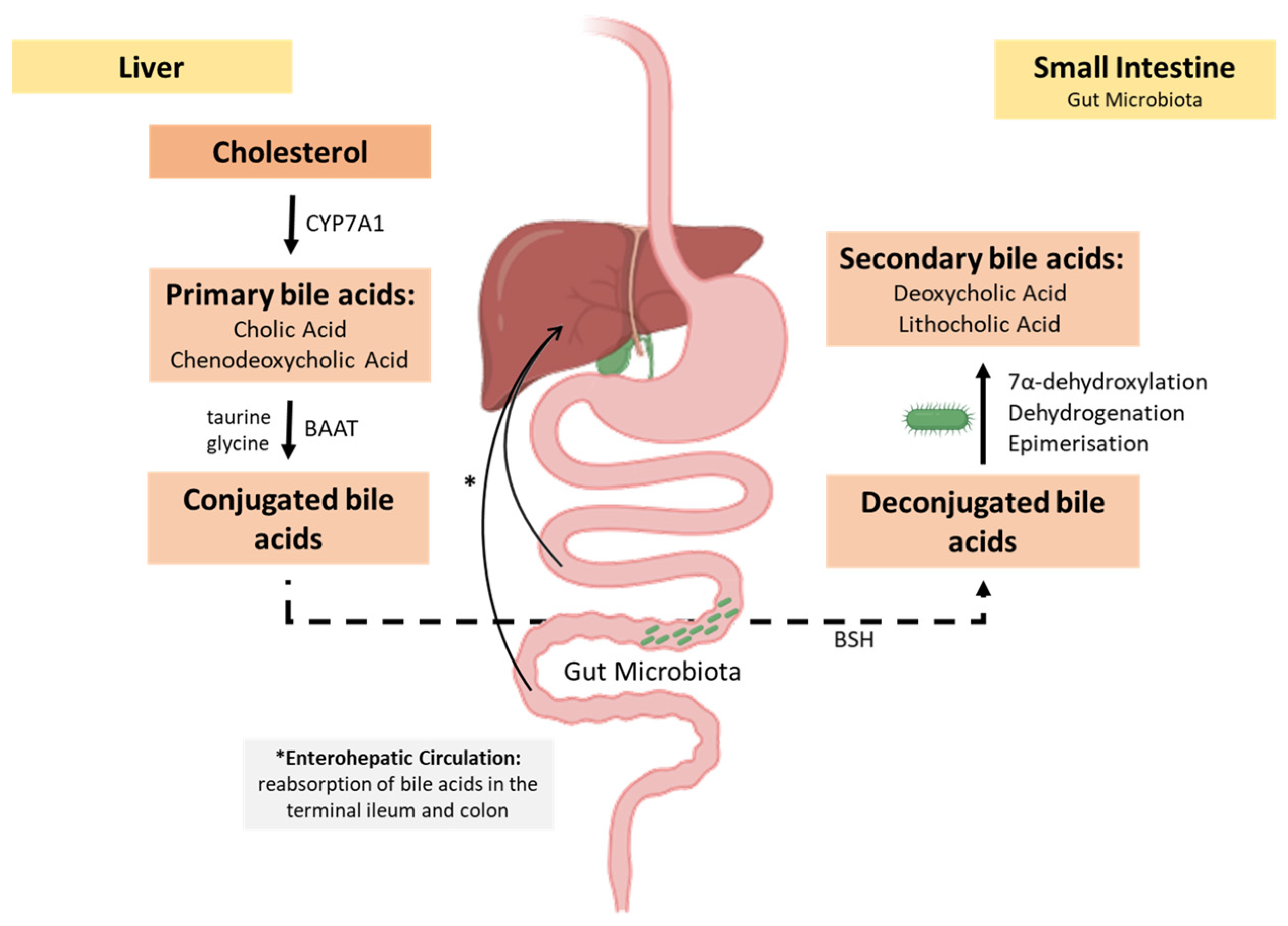
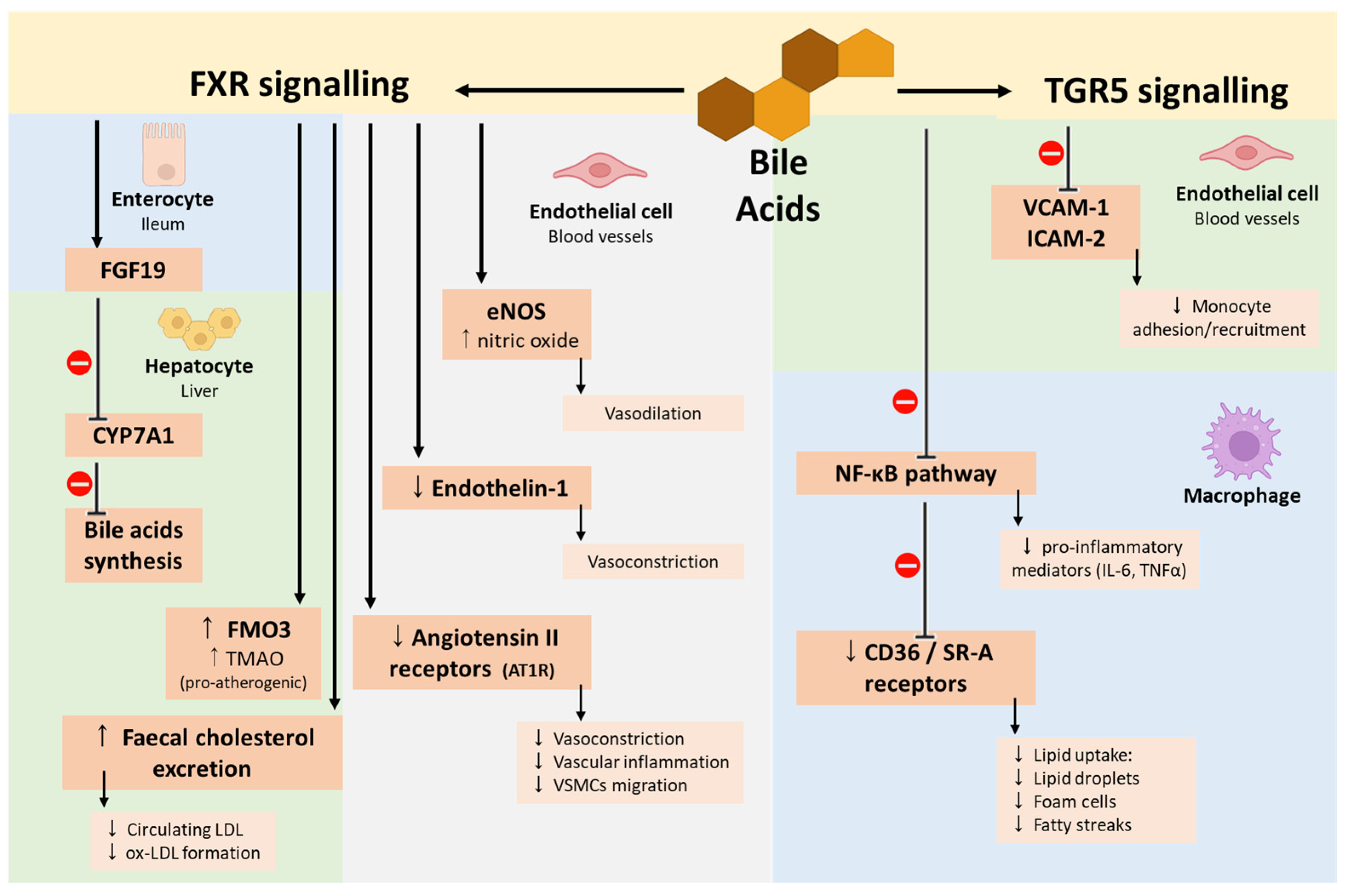
4.2. Bile Acids
4.3. Short-Chain Fatty Acids
4.4. Tryptophan Metabolites
4.5. Non-Bacterial Microbiota and Atherosclerosis
4.6. Microbiota-Induced Immune and Inflammatory Pathways in Atherosclerosis
5. Gut Microbiota-Targeted Interventions in the Prevention and Progression of Atherosclerosis
5.1. Prebiotics
5.2. Probiotics
5.3. Faecal Microbiota Transplantation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barquera, S.; Pedroza-Tobias, A.; Medina, C.; Hernandez-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; David, S.G.; David, A.G.; Tarca, V.; Paduret, I.A.; Mindru, D.E.; Rosu, S.T.; Rosu, E.V.; Adumitrachioaiei, H.; Bernic, J.; et al. Atherosclerosis from newborn to adult-epidemiology, pathological aspects, and risk factors. Life 2023, 13, 2056. [Google Scholar] [CrossRef]
- Papadopoulos, P.D.; Tsigalou, C.; Valsamaki, P.N.; Konstantinidis, T.G.; Voidarou, C.; Bezirtzoglou, E. The emerging role of the gut microbiome in cardiovascular disease: Current knowledge and perspectives. Biomedicines 2022, 10, 948. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fak, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 4592–4598. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Del Campo-Moreno, R.; Alarcon-Cavero, T.; D’Auria, G.; Delgado-Palacio, S.; Ferrer-Martinez, M. Microbiota and human health: Characterization techniques and transference. Enferm. Infecc. Microbiol. Clin. (Engl. Ed.) 2018, 36, 241–245. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Sniffen, S.; McGill Percy, K.C.; Pallaval, V.B.; Chidipi, B. Gut dysbiosis and immune system in atherosclerotic cardiovascular disease (ACVD). Microorganisms 2022, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut microbiota in hypertension and atherosclerosis: A review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef]
- Almeida, C.; Barata, P.; Fernandes, R. The influence of gut microbiota in cardiovascular diseases-a brief review. Porto Biomed. J. 2021, 6, e106. [Google Scholar] [CrossRef]
- Dimri, S.; Rizzo, A. The Gut Microbiome: An overview. J. Stud. Res. 2022, 11, 1–14. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, F.; Harun-Or-Rashid, M.; Mamun, A.A.; Rahaman, M.S.; Islam, M.M.; Meem, A.F.K.; Sutradhar, P.R.; Mitra, S.; Mimi, A.A.; et al. The gut microbiota (microbiome) in cardiovascular disease and its therapeutic regulation. Front. Cell. Infect. Microbiol. 2022, 12, 903570. [Google Scholar] [CrossRef] [PubMed]
- Matijasic, M.; Mestrovic, T.; Paljetak, H.C.; Peric, M.; Baresic, A.; Verbanac, D. Gut microbiota beyond bacteria-mycobiome, virome, archaeome, and eukaryotic parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Lee, K.T. Atherosclerosis by virus infection-A short review. Biomedicines 2022, 10, 2634. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, J.; Meng, J.; Li, S.; Zhang, Y.; You, W.; Sai, X.; Yang, J.; Zhang, S.; Sun, W. Structural changes in the gut virome of patients with atherosclerotic cardiovascular disease. Microbiol. Spectr. 2024, 12, e0105023. [Google Scholar] [CrossRef]
- El Jaddaoui, I.; Sehli, S.; Al Idrissi, N.; Bakri, Y.; Belyamani, L.; Ghazal, H. The gut mycobiome for precision medicine. J. Fungi 2025, 11, 279. [Google Scholar] [CrossRef]
- Buttar, J.; Kon, E.; Lee, A.; Kaur, G.; Lunken, G. Effect of diet on the gut mycobiome and potential implications in inflammatory bowel disease. Gut Microbes 2024, 16, 2399360. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, S.; Hu, X.; Ye, C.; Nie, Q.; Wang, K.; Yan, S.; Lin, J.; Xu, F.; Li, M.; et al. Candida albicans accelerates atherosclerosis by activating intestinal hypoxia-inducible factor 2-alpha signaling. Cell Host Microbe 2024, 32, 964–979.e7. [Google Scholar] [CrossRef]
- Dieterich, W.; Schink, M.; Zopf, Y. Microbiota in the gastrointestinal tract. Med. Sci. 2018, 6, 116. [Google Scholar] [CrossRef]
- Di Vincenzo, F.; Del Gaudio, A.; Petito, V.; Lopetuso, L.R.; Scaldaferri, F. Gut microbiota, intestinal permeability, and systemic inflammation: A narrative review. Intern. Emerg. Med. 2024, 19, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Afzaal, M.; Saeed, F.; Shah, Y.A.; Hussain, M.; Rabail, R.; Socol, C.T.; Hassoun, A.; Pateiro, M.; Lorenzo, J.M.; Rusu, A.V.; et al. Human gut microbiota in health and disease: Unveiling the relationship. Front. Microbiol. 2022, 13, 999001. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Fernandez Real, J.M.; Guarner, F.; Gueimonde, M.; Rodriguez, J.M.; Saenz de Pipaon, M.; Sanz, Y. Gut microbes and health. Gastroenterol. Hepatol. 2021, 44, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Hrncir, T. Gut microbiota dysbiosis: Triggers, consequences, diagnostic and therapeutic options. Microorganisms 2022, 10, 578. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Singh, R.; Ro, S.; Ghoshal, U.C. Gut microbiota dysbiosis in functional gastrointestinal disorders: Underpinning the symptoms and pathophysiology. JGH Open 2021, 5, 976–987. [Google Scholar] [CrossRef]
- Saeed, N.K.; Al-Beltagi, M.; Bediwy, A.S.; El-Sawaf, Y.; Toema, O. Gut microbiota in various childhood disorders: Implication and indications. World J. Gastroenterol. 2022, 28, 1875–1901. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Vemuri, R.; Gundamaraju, R.; Shastri, M.D.; Shukla, S.D.; Kalpurath, K.; Ball, M.; Tristram, S.; Shankar, E.M.; Ahuja, K.; Eri, R. Gut microbial changes, interactions, and their implications on human lifecycle: An ageing perspective. Biomed. Res. Int. 2018, 2018, 4178607. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, Z.; Zhang, W.; Zhang, C.; Zhang, Y.; Mei, H.; Zhuo, N.; Wang, H.; Wang, L.; Wu, D. Comparison of gut microbiota in exclusively breast-fed and formula-fed babies: A study of 91 term infants. Sci. Rep. 2020, 10, 15792. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.A.; Iyengar, R.S. Breast milk, microbiota, and intestinal immune homeostasis. Pediatr. Res. 2015, 77, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Homann, C.M.; Rossel, C.A.J.; Dizzell, S.; Bervoets, L.; Simioni, J.; Li, J.; Gunn, E.; Surette, M.G.; de Souza, R.J.; Mommers, M.; et al. Infants’ first solid foods: Impact on gut microbiota development in two intercontinental cohorts. Nutrients 2021, 13, 2639. [Google Scholar] [CrossRef]
- Differding, M.K.; Benjamin-Neelon, S.E.; Hoyo, C.; Ostbye, T.; Mueller, N.T. Timing of complementary feeding is associated with gut microbiota diversity and composition and short chain fatty acid concentrations over the first year of life. BMC Microbiol. 2020, 20, 56. [Google Scholar] [CrossRef] [PubMed]
- Fewtrell, M.; Bronsky, J.; Campoy, C.; Domellof, M.; Embleton, N.; Fidler Mis, N.; Hojsak, I.; Hulst, J.M.; Indrio, F.; Lapillonne, A.; et al. Complementary Feeding: A Position Paper by the European Society for Paediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) Committee on Nutrition. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [Google Scholar] [CrossRef]
- Sarkar, A.; Yoo, J.Y.; Valeria Ozorio Dutra, S.; Morgan, K.H.; Groer, M. The Association between Early-Life Gut Microbiota and Long-Term Health and Diseases. J. Clin. Med. 2021, 10, 459. [Google Scholar] [CrossRef]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef]
- Wu, W.M.; Yang, Y.S.; Peng, L.H. Microbiota in the stomach: New insights. J. Dig. Dis. 2014, 15, 54–61. [Google Scholar] [CrossRef]
- Rajilic-Stojanovic, M.; Figueiredo, C.; Smet, A.; Hansen, R.; Kupcinskas, J.; Rokkas, T.; Andersen, L.; Machado, J.C.; Ianiro, G.; Gasbarrini, A.; et al. Systematic review: Gastric microbiota in health and disease. Aliment. Pharmacol. Ther. 2020, 51, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Compare, D. The human gastric microbiota: Is it time to rethink the pathogenesis of stomach diseases? United Eur. Gastroenterol. J. 2015, 3, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.A.H.; Heyndrickx, M.; Jonkers, D.; Mackie, A.; Millet, S.; Naghibi, M.; Paerregaard, S.I.; Pot, B.; Saulnier, D.; Sina, C.; et al. Small intestine vs. colon ecology and physiology: Why it matters in probiotic administration. Cell Rep. Med. 2023, 4, 101190. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; van den Bogert, B.; Kleerebezem, M. The small intestine microbiota, nutritional modulation and relevance for health. Curr. Opin. Biotechnol. 2015, 32, 14–20. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Zheng, L.; Kelly, C.J.; Colgan, S.P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C350–C360. [Google Scholar] [CrossRef]
- Villmones, H.C.; Svanevik, M.; Ulvestad, E.; Stenstad, T.; Anthonisen, I.L.; Nygaard, R.M.; Dyrhovden, R.; Kommedal, O. Investigating the human jejunal microbiota. Sci. Rep. 2022, 12, 1682. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Villmones, H.C.; Haug, E.S.; Ulvestad, E.; Grude, N.; Stenstad, T.; Halland, A.; Kommedal, O. Species level description of the human ileal bacterial microbiota. Sci. Rep. 2018, 8, 4736. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Matzaras, R.; Nikopoulou, A.; Protonotariou, E.; Christaki, E. Gut microbiota modulation and prevention of dysbiosis as an alternative approach to antimicrobial resistance: A narrative review. Yale J. Biol. Med. 2022, 95, 479–494. [Google Scholar] [PubMed]
- İlhan, N. Gut microbiota and metabolism. Int. J. Med. Biochem. 2018, 1, 115–128. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, Y.; Cheng, H.; Zhang, D.; Feng, W.; Peng, C. Functions of gut microbiota metabolites, current status and future perspectives. Aging Dis. 2022, 13, 1106–1126. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Alexandrescu, L.; Suceveanu, A.P.; Stanigut, A.M.; Tofolean, D.E.; Axelerad, A.D.; Iordache, I.E.; Herlo, A.; Nelson Twakor, A.; Nicoara, A.D.; Tocia, C.; et al. Intestinal insights: The gut microbiome’s role in atherosclerotic disease: A narrative review. Microorganisms 2024, 12, 2341. [Google Scholar] [CrossRef]
- Salamone, D.; Rivellese, A.A.; Vetrani, C. The relationship between gut microbiota, short-chain fatty acids and type 2 diabetes mellitus: The possible role of dietary fibre. Acta Diabetol. 2021, 58, 1131–1138. [Google Scholar] [CrossRef]
- Shanmugham, M.; Bellanger, S.; Leo, C.H. Gut-derived metabolite, trimethylamine-N-oxide (TMAO) in cardio-metabolic diseases: Detection, mechanism, and potential therapeutics. Pharmaceuticals 2023, 16, 504. [Google Scholar] [CrossRef]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 2015, 6, e02481. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Cespedes, I.; Bennett, B.J.; Allayee, H. Expanding role of gut microbiota in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Chen, C.; Li, P.; Lu, B. The gut microbiota-artery axis: A bridge between dietary lipids and atherosclerosis? Prog. Lipid Res. 2023, 89, 101209. [Google Scholar] [CrossRef] [PubMed]
- Hadadi, N.; Berweiler, V.; Wang, H.; Trajkovski, M. Intestinal microbiota as a route for micronutrient bioavailability. Curr. Opin. Endocr. Metab. Res. 2021, 20, 100285. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front. Nutr. 2019, 6, 48. [Google Scholar] [CrossRef]
- Hertli, S.; Zimmermann, P. Molecular interactions between the intestinal microbiota and the host. Mol. Microbiol. 2022, 117, 1297–1307. [Google Scholar] [CrossRef]
- Belkaid, Y.; Harrison, O.J. Homeostatic immunity and the microbiota. Immunity 2017, 46, 562–576. [Google Scholar] [CrossRef]
- Choden, T.; Cohen, N.A. The gut microbiome and the immune system. Explor. Med. 2022, 3, 219–233. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Cruz Neto, J.P.R.; de Luna Freire, M.O.; de Albuquerque Lemos, D.E.; Ribeiro Alves, R.M.F.; de Farias Cardoso, E.F.; de Moura Balarini, C.; Duman, H.; Karav, S.; de Souza, E.L.; de Brito Alves, J.L. Targeting gut microbiota with probiotics and phenolic compounds in the treatment of atherosclerosis: A comprehensive review. Foods 2024, 13, 2886. [Google Scholar] [CrossRef]
- Jergens, A.E.; Parvinroo, S.; Kopper, J.; Wannemuehler, M.J. Rules of engagement: Epithelial-microbe interactions and inflammatory bowel disease. Front. Med. 2021, 8, 669913. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Leaky gut: Mechanisms, measurement and clinical implications in humans. Gut 2019, 68, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Chevalier-Curt, M.J.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.K.; Azmal, M.; Haque, A.; Meem, M.; Talukder, O.F.; Ghosh, A. Unlocking the secrets of the human gut microbiota: Comprehensive review on its role in different diseases. World J. Gastroenterol. 2025, 31, 99913. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert. Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Venegas, D.P.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Mousa, W.K.; Chehadeh, F.; Husband, S. Microbial dysbiosis in the gut drives systemic autoimmune diseases. Front. Immunol. 2022, 13, 906258. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Mendez-Garcia, C.; Rojo, D.; Barbas, C.; Moya, A. Antibiotic use and microbiome function. Biochem. Pharmacol. 2017, 134, 114–126. [Google Scholar] [CrossRef]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 axis: A dynamic balance regulated by the gut microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as major disruptors of gut microbiota. Front. Cell Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef] [PubMed]
- Demehri, F.R.; Barrett, M.; Teitelbaum, D.H. Changes to the intestinal microbiome with parenteral nutrition: Review of a murine model and potential clinical implications. Nutr. Clin. Pract. 2015, 30, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Magee, E.A.; Richardson, C.J.; Hughes, R.; Cummings, J.H. Contribution of dietary protein to sulfide production in the large intestine: An in vitro and a controlled feeding study in humans. Am. J. Clin. Nutr. 2000, 72, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef]
- Moreira de Gouveia, M.I.; Bernalier-Donadille, A.; Jubelin, G. Enterobacteriaceae in the human gut: Dynamics and ecological roles in health and disease. Biology 2024, 13, 142. [Google Scholar] [CrossRef]
- Gusarov, I.; Shatalin, K.; Starodubtseva, M.; Nudler, E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 2009, 325, 1380–1384. [Google Scholar] [CrossRef]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The impact of oxidative stress in human pathology: Focus on gastrointestinal disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- Swiatecka, D.; Narbad, A.; Ridgway, K.P.; Kostyra, H. The study on the impact of glycated pea proteins on human intestinal bacteria. Int. J. Food Microbiol. 2011, 145, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Chen, S.; Lin, L. Research progress of gut microbiota and obesity caused by high-fat diet. Front. Cell Infect. Microbiol. 2023, 13, 1139800. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.S. Changes seen in gut bacteria content and distribution with obesity: Causation or association? Postgrad. Med. 2015, 127, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Yokota, A.; Fukiya, S.; Islam, K.B.; Ooka, T.; Ogura, Y.; Hayashi, T.; Hagio, M.; Ishizuka, S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes 2012, 3, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, R.; Inoue, K.Y.; Nishino, K.; Yamasaki, S. Intestinal and fecal pH in human health. Front. Microbiomes 2023, 2, 1192316. [Google Scholar] [CrossRef]
- Natividad, J.M.; Lamas, B.; Pham, H.P.; Michel, M.L.; Rainteau, D.; Bridonneau, C.; da Costa, G.; van Hylckama Vlieg, J.; Sovran, B.; Chamignon, C.; et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat. Commun. 2018, 9, 2802. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, T.; Gu, Y.; Wang, X.; Xie, R.; Sun, Y.; Wang, B.; Cao, H. Regulation of gut microbiota-bile acids axis by probiotics in inflammatory bowel disease. Front. Immunol. 2022, 13, 974305. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Pang, X.; Zhao, Y.; Wang, L.; Zhao, L. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations. ISME J. 2012, 6, 1848–1857. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The gut microbiota and inflammation: An overview. Int. J. Environ. Res. Public. Health 2020, 17, 7681. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, Z.; Li, M.D. Effect of cigarette Smoke on gut microbiota: State of knowledge. Front. Physiol. 2021, 12, 673341. [Google Scholar] [CrossRef]
- Rojas-Valverde, D.; Bonilla, D.A.; Gomez-Miranda, L.M.; Calleja-Nunez, J.J.; Arias, N.; Martinez-Guardado, I. Examining the interaction between exercise, gut microbiota, and neurodegeneration: Future research directions. Biomedicines 2023, 11, 2267. [Google Scholar] [CrossRef] [PubMed]
- Boytar, A.N.; Skinner, T.L.; Wallen, R.E.; Jenkins, D.G.; Dekker Nitert, M. The effect of exercise prescription on the human gut microbiota and comparison between clinical and apparently healthy populations: A systematic review. Nutrients 2023, 15, 1534. [Google Scholar] [CrossRef] [PubMed]
- Dziewiecka, H.; Buttar, H.S.; Kasperska, A.; Ostapiuk-Karolczuk, J.; Domagalska, M.; Cichon, J.; Skarpanska-Stejnborn, A. Physical activity induced alterations of gut microbiota in humans: A systematic review. BMC Sports Sci. Med. Rehabil. 2022, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, W.; Liu, W.; Zhou, M. Roles and mechanisms of renalase in cardiovascular disease: A promising therapeutic target. Biomed. Pharmacother. 2020, 131, 110712. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 12 August 2024).
- Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Makaryus, A.N.; Wengrofsky, P.; Lee, J. Dyslipidemia and its role in the pathogenesis of atherosclerotic cardiovascular disease: Implications for evaluation and targets for treatment of dyslipidemia based on recent guidelines. In Dyslipidemia; McFarlane, S.I., Ed.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Lu, Y.; Cui, X.; Zhang, L.; Wang, X.; Xu, Y.; Qin, Z.; Liu, G.; Wang, Q.; Tian, K.; Lim, K.S.; et al. The functional role of lipoproteins in atherosclerosis: Novel directions for diagnosis and targeting therapy. Aging Dis. 2022, 13, 491–520. [Google Scholar] [CrossRef]
- Bjorkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular endothelial cell biology: An update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Alexander, Y.; Osto, E.; Schmidt-Trucksass, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Back, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Lv, Y.; Bai, X.; Qi, J.; Weng, X.; Liu, S.; Bao, X.; Jia, H.; Yu, B. Plaque erosion: A distinctive pathological mechanism of acute coronary syndrome. Front. Cardiovasc. Med. 2021, 8, 711453. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, S.; Li, J.; Wang, L.; Zhang, Q.; Hou, L.; Yu, X.; Liu, Z.; Lv, T.; Shang, L. Low shear stress induces vascular endothelial cells apoptosis via miR-330 /SOD2 /HSP70 signaling pathway. Exp. Cell Res. 2025, 445, 114410. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef]
- Higashi, Y. Endothelial function in dyslipidemia: Roles of LDL-cholesterol, HDL-cholesterol and triglycerides. Cells 2023, 12, 1293. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic insights into the oxidized low-density lipoprotein-induced atherosclerosis. Oxid. Med. Cell Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Roy, P.; Orecchioni, M.; Ley, K. How the immune system shapes atherosclerosis: Roles of innate and adaptive immunity. Nat. Rev. Immunol. 2022, 22, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Ahmed, S.F., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam cells in atherosclerosis: Novel insights into its origins, consequences, and molecular mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arter. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef]
- Peled, M.; Fisher, E.A. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front. Immunol. 2014, 5, 579. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Ortega, C.; Francis, G.A. Smooth Muscle Cell-Proteoglycan-Lipoprotein Interactions as Drivers of Atherosclerosis. In Prevention and Treatment of Atherosclerosis: Improving State-of-the-Art Management and Search for Novel Targets; von Eckardstein, A., Binder, C.J., Eds.; Springer: Cham, Switzerland, 2022; pp. 335–358. [Google Scholar]
- Beck-Joseph, J.; Tabrizian, M.; Lehoux, S. Molecular Interactions Between Vascular Smooth Muscle Cells and Macrophages in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 737934. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Vergallo, R.; Crea, F. Atherosclerotic plaque healing. N. Engl. J. Med. 2020, 383, 846–857. [Google Scholar] [CrossRef]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arter. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Monaji, S.R. Steps of atherosclerosis and its complication. J. Med. Pharm. Allied Sci. 2020, 9, 2577–2579. [Google Scholar] [CrossRef]
- Hermiz, C.; Sedhai, Y.R. Angina. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Takahashi, J.; Onuma, S.; Hao, K.; Godo, S.; Shiroto, T.; Yasuda, S. Pathophysiology and diagnostic pathway of myocardial infarction with non-obstructive coronary arteries. J. Cardiol. 2024, 83, 17–24. [Google Scholar] [CrossRef]
- Salaudeen, M.A.; Bello, N.; Danraka, R.N.; Ammani, M.L. Understanding the pathophysiology of ischemic stroke: The basis of current therapies and opportunity for new ones. Biomolecules 2024, 14, 305. [Google Scholar] [CrossRef]
- Murphy, K.; O’Donovan, A.N.; Caplice, N.M.; Ross, R.P.; Stanton, C. Exploring the gut microbiota and cardiovascular disease. Metabolites 2021, 11, 493. [Google Scholar] [CrossRef]
- Eshghjoo, S.; Jayaraman, A.; Sun, Y.; Alaniz, R.C. Microbiota-mediated immune regulation in atherosclerosis. Molecules 2021, 26, 179. [Google Scholar] [CrossRef] [PubMed]
- Luqman, A.; Hassan, A.; Ullah, M.; Naseem, S.; Ullah, M.; Zhang, L.; Din, A.U.; Ullah, K.; Ahmad, W.; Wang, G. Role of the intestinal microbiome and its therapeutic intervention in cardiovascular disorder. Front. Immunol. 2024, 15, 1321395. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.K. Role of gut microbiota and their metabolites on atherosclerosis, hypertension and human blood platelet function: A review. Nutrients 2021, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the gap between gut microbial dysbiosis and cardiovascular diseases. Nutrients 2017, 9, 859. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Huang, P.; Liu, D.; Xing, G.; Guo, R.; Li, S.; Fan, S.; Cheng, L.; Yan, Q.; Yang, W. Gut mycobiome alterations and network interactions with the bacteriome in patients with atherosclerotic cardiovascular disease. Microbiol. Spectr. 2025, 13, e0218224. [Google Scholar] [CrossRef]
- An, K.; Jia, Y.; Xie, B.; Gao, J.; Chen, Y.; Yuan, W.; Zhong, J.; Su, P.; Liu, X. Alterations in the gut mycobiome with coronary artery disease severity. EBioMedicine 2024, 103, 105137. [Google Scholar] [CrossRef] [PubMed]
- Kirk, D.; Costeira, R.; Visconti, A.; Khan Mirzaei, M.; Deng, L.; Valdes, A.M.; Menni, C. Bacteriophages, gut bacteria, and microbial pathways interplay in cardiometabolic health. Cell Rep. 2024, 43, 113728. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Q.; Jiang, H. Gut microbiota in atherosclerosis: Focus on trimethylamine N-oxide. APMIS 2020, 128, 353–366. [Google Scholar] [CrossRef]
- Canyelles, M.; Borras, C.; Rotllan, N.; Tondo, M.; Escola-Gil, J.C.; Blanco-Vaca, F. Gut microbiota-derived TMAO: A causal factor promoting atherosclerotic cardiovascular disease? Int. J. Mol. Sci. 2023, 24, 1940. [Google Scholar] [CrossRef]
- Papandreou, C.; More, M.; Bellamine, A. Trimethylamine N-oxide in relation to cardiometabolic health-cause or effect? Nutrients 2020, 12, 1330. [Google Scholar] [CrossRef]
- Zhen, J.; Zhou, Z.; He, M.; Han, H.X.; Lv, E.H.; Wen, P.B.; Liu, X.; Wang, Y.T.; Cai, X.C.; Tian, J.Q.; et al. The gut microbial metabolite trimethylamine N-oxide and cardiovascular diseases. Front. Endocrinol. 2023, 14, 1085041. [Google Scholar] [CrossRef] [PubMed]
- Querio, G.; Antoniotti, S.; Geddo, F.; Levi, R.; Gallo, M.P. Modulation of endothelial function by TMAO, a gut microbiota-derived metabolite. Int. J. Mol. Sci. 2023, 24, 5806. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.B.; Zhang, Y.; Boini, K.M.; Koka, S. High mobility group box 1 mediates TMAO-induced endothelial dysfunction. Int. J. Mol. Sci. 2019, 20, 3570. [Google Scholar] [CrossRef]
- Holme, S.A.N.; Sigsgaard, T.; Holme, J.A.; Holst, G.J. Effects of particulate matter on atherosclerosis: A link via high-density lipoprotein (HDL) functionality? Part. Fibre Toxicol. 2020, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yu, X.; Wang, C.; Zhao, H.; Wang, X.; Guan, X. Trimethylamine N-oxide promotes oxidative stress and lipid accumulation in macrophage foam cells via the Nrf2/ABCA1 pathway. J. Physiol. Biochem. 2024, 80, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Zhang, H.; Xiang, Q.; Shen, L.; Guo, X.; Zhai, C.; Hu, H. Targeting trimethylamine N-Oxide: A new therapeutic strategy for alleviating atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 864600. [Google Scholar] [CrossRef]
- Vourakis, M.; Mayer, G.; Rousseau, G. The role of gut microbiota on cholesterol metabolism in atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8074. [Google Scholar] [CrossRef]
- Xu, W.; Zou, K.; Zhan, Y.; Cai, Y.; Zhang, Z.; Tao, X.; Qiu, L.; Wei, H. Enterococcus faecium GEFA01 alleviates hypercholesterolemia by promoting reverse cholesterol transportation via modulating the gut microbiota-SCFA axis. Front. Nutr. 2022, 9, 1020734. [Google Scholar] [CrossRef]
- Witkowski, M.; Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; et al. Vascular endothelial tissue factor contributes to trimethylamine N-oxide-enhanced arterial thrombosis. Cardiovasc. Res. 2022, 118, 2367–2384. [Google Scholar] [CrossRef]
- Wang, B.; Qiu, J.; Lian, J.; Yang, X.; Zhou, J. Gut metabolite trimethylamine-N-oxide in atherosclerosis: From mechanism to therapy. Front. Cardiovasc. Med. 2021, 8, 723886. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Perez, O.; Cruz-Ramon, V.; Chinchilla-Lopez, P.; Mendez-Sanchez, N. The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 2017, 16 (Suppl. S1), S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Majait, S.; Nieuwdorp, M.; Kemper, M.; Soeters, M. The black box orchestra of gut bacteria and bile acids: Who is the conductor? Int. J. Mol. Sci. 2023, 24, 1816. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Morillo, V.; Suarez, M.K.; Castro, A.; Ramirez, P.; Rojas, M.; Anez, R.; D’Marco, L.; Chacin-Gonzalez, M.; Bermudez, V. Role of gut microbiome in atherosclerosis: Molecular and therapeutic aspects. Curr. Cardiol. Rev. 2023, 19, e020223213408. [Google Scholar] [CrossRef]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap-bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Yntema, T.; Koonen, D.P.Y.; Kuipers, F. Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases. Nutrients 2023, 15, 1850. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Yi, Q.; Luo, L.; Xiong, Y. Regulation of bile acids and their receptor FXR in metabolic diseases. Front. Nutr. 2024, 11, 1447878. [Google Scholar] [CrossRef]
- Xu, Y.; Li, F.; Zalzala, M.; Xu, J.; Gonzalez, F.J.; Adorini, L.; Lee, Y.K.; Yin, L.; Zhang, Y. Farnesoid X receptor activation increases reverse cholesterol transport by modulating bile acid composition and cholesterol absorption in mice. Hepatology 2016, 64, 1072–1085. [Google Scholar] [CrossRef]
- Singh, A.B.; Dong, B.; Kraemer, F.B.; Xu, Y.; Zhang, Y.; Liu, J. Farnesoid X receptor activation by obeticholic acid elevates liver low-density lipoprotein receptor expression by mRNA stabilization and reduces plasma low-density lipoprotein cholesterol in mice. Arter. Thromb. Vasc. Biol. 2018, 38, 2448–2459. [Google Scholar] [CrossRef]
- Ma, J.; Li, H. The role of gut microbiota in atherosclerosis and hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, J.; Wu, W.; Zhu, Y.; Liu, X. The role of bile acids in cardiovascular diseases: From mechanisms to clinical implications. Aging Dis. 2023, 14, 261–282. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M.; Wu, Y.; Boehme, S. Bile acid and cholesterol metabolism in atherosclerotic cardiovascular disease and therapy. Cardiol. Plus 2020, 5, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, M.; Rout, A.; Kingsley, T.; Kirchoff, R.; Singh, A.; Verma, V.; Kant, R.; Chaudhary, R. Role of gut microbiota in cardiovascular diseases. World J. Cardiol. 2020, 12, 110–122. [Google Scholar] [CrossRef]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Ao, H.; Peng, C. Gut microbiota, short-chain fatty acids, and herbal medicines. Front. Pharmacol. 2018, 9, 1354. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.P.; Chapman, J.A.; Stewart, C.J. Gut microbiome derived short chain fatty acids: Promising strategies in necrotising enterocolitis. Curr. Res. Microb. Sci. 2024, 6, 100219. [Google Scholar] [CrossRef] [PubMed]
- Deleu, S.; Machiels, K.; Raes, J.; Verbeke, K.; Vermeire, S. Short chain fatty acids and its producing organisms: An overlooked therapy for IBD? EBioMedicine 2021, 66, 103293. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A double-edged sword for health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Salazar, N. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; McWilliam Leitch, C.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, Y.; Li, X.; Sun, B. Dynamic balancing of intestinal short-chain fatty acids: The crucial role of bacterial metabolism. Trends Food Sci. Technol. 2020, 100, 118–130. [Google Scholar] [CrossRef]
- Richards, L.B.; Li, M.; van Esch, B.C.A.M.; Garssen, J.; Folkerts, G. The effects of short-chain fatty acids on the cardiovascular system. PharmaNutrition 2016, 4, 68–111. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef] [PubMed]
- Zapolska-Downar, D.; Siennicka, A.; Kaczmarczyk, M.; Kolodziej, B.; Naruszewicz, M. Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: The role of NF-kappaB and PPARalpha. J. Nutr. Biochem. 2004, 15, 220–228. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.; Henricks, P.A.J.; Folkerts, G.; Garssen, J. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide or tumor necrosis factor alpha-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef]
- Feng, Y.; Xu, D. Short-chain fatty acids are potential goalkeepers of atherosclerosis. Front. Pharmacol. 2023, 14, 1271001. [Google Scholar] [CrossRef]
- Aguilar, E.C.; Leonel, A.J.; Teixeira, L.G.; Silva, A.R.; Silva, J.F.; Pelaez, J.M.; Capettini, L.S.; Lemos, V.S.; Santos, R.A.; Alvarez-Leite, J.I. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFkappaB activation. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 606–613. [Google Scholar] [CrossRef]
- Xiao, Y.; Guo, Z.; Li, Z.; Ling, H.; Song, C. Role and mechanism of action of butyrate in atherosclerotic diseases: A review. J. Appl. Microbiol. 2021, 131, 543–552. [Google Scholar] [CrossRef]
- Mao, Y.; Kong, C.; Zang, T.; You, L.; Wang, L.S.; Shen, L.; Ge, J.B. Impact of the gut microbiome on atherosclerosis. mLife 2024, 3, 167–175. [Google Scholar] [CrossRef]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Paeslack, N.; Mimmler, M.; Becker, S.; Gao, Z.; Khuu, M.P.; Mann, A.; Malinarich, F.; Regen, T.; Reinhardt, C. Microbiota-derived tryptophan metabolites in vascular inflammation and cardiovascular disease. Amino Acids 2022, 54, 1339–1356. [Google Scholar] [CrossRef]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van Winkle, L.; Matsumura, F.; Vogel, C.F. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arter. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef]
- Xue, H.; Chen, X.; Yu, C.; Deng, Y.; Zhang, Y.; Chen, S.; Chen, X.; Chen, K.; Yang, Y.; Ling, W. Gut Microbially Produced Indole-3-Propionic Acid Inhibits Atherosclerosis by Promoting Reverse Cholesterol Transport and Its Deficiency Is Causally Related to Atherosclerotic Cardiovascular Disease. Circ. Res. 2022, 131, 404–420. [Google Scholar] [CrossRef]
- Chuang, H.L.; Chiu, C.C.; Lo, C.; Hsu, C.C.; Liu, J.Y.; Hung, S.W.; Tsai, S.C.; Sung, H.H.; Wang, C.L.; Huang, Y.T. Circulating gut microbiota-related metabolites influence endothelium plaque lesion formation in ApoE knockout rats. PLoS ONE 2022, 17, e0264934. [Google Scholar] [CrossRef]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Pawlak, D. Indoxyl sulfate—The uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef]
- Hung, S.C.; Kuo, K.L.; Wu, C.C.; Tarng, D.C. Indoxyl sulfate: A novel cardiovascular risk factor in chronic kidney disease. J. Am. Heart Assoc. 2017, 6, e005022. [Google Scholar] [CrossRef]
- Skye, S.M.; Hazen, S.L. Microbial modulation of a uremic toxin. Cell Host Microbe 2016, 20, 691–692. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Burtey, S.; Sallee, M. Indoxyl sulfate, a uremic endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Aschenbrenner, D.; Yoo, J.Y.; Zuo, T. The gut mycobiome in health, disease, and clinical applications in association with the gut bacterial microbiome assembly. Lancet Microbe 2022, 3, e969–e983. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, B.S.; Rosu, O.A.; Enache, R.M.; Profir, M.M.; Pavelescu, L.A.; Cretoiu, S.M. Gut mycobiome: Latest findings and current knowledge regarding its significance in human health and disease. J. Fungi 2025, 11, 333. [Google Scholar] [CrossRef]
- Chacon, M.R.; Lozano-Bartolome, J.; Portero-Otin, M.; Rodriguez, M.M.; Xifra, G.; Puig, J.; Blasco, G.; Ricart, W.; Chaves, F.J.; Fernandez-Real, J.M. The gut mycobiome composition is linked to carotid atherosclerosis. Benef. Microbes 2018, 9, 185–198. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Shen, X.; Li, L.; Sun, Z.; Zang, G.; Zhang, L.; Shao, C.; Wang, Z. Gut microbiota and atherosclerosis-focusing on the plaque stability. Front. Cardiovasc. Med. 2021, 8, 668532. [Google Scholar] [CrossRef]
- Takiishi, T.; Fenero, C.I.M.; Camara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef]
- Parantainen, J.; Barreto, G.; Strandberg, T.E.; Mars, N.; Nurmi, K.; Eklund, K.K. Increased intestinal mucosal permeability and metabolic endotoxemia predict the risk of cardiovascular mortality. Atherosclerosis 2025, 405, 119220. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Wick, G.; Jakic, B.; Buszko, M.; Wick, M.C.; Grundtman, C. The role of heat shock proteins in atherosclerosis. Nat. Rev. Cardiol. 2014, 11, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Metzler, B.; Jahangiri, M.; Mandal, K. Molecular chaperones and heat shock proteins in atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H506–H514. [Google Scholar] [CrossRef] [PubMed]
- Falck-Hansen, M.; Kassiteridi, C.; Monaco, C. Toll-like receptors in atherosclerosis. Int. J. Mol. Sci. 2013, 14, 14008–14023. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V. Regulation of nitrate and nitrite reductase synthesis in enterobacteria. Antonie Van Leeuwenhoek 1994, 66, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Xu, D. Research progress on Th17 and T regulatory cells and their cytokines in regulating atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 929078. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Frutos Rde, L.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef]
- Wang, Y.; Zang, J.; Liu, C.; Yan, Z.; Shi, D. Interleukin-17 links Inflammatory cross-talks between comorbid psoriasis and atherosclerosis. Front. Immunol. 2022, 13, 835671. [Google Scholar] [CrossRef]
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; Stellos, K.; Little, K.M.; Lasitschka, F.; Doesch, A.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol. 2014, 193, 4344–4355. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Liu, X.; Awar, L.; McMickle, A.; Bai, F.; Nagarajan, S.; Yu, S. IL-17 induces expression of vascular cell adhesion molecule through signalling pathway of NF-kappaB, but not Akt1 and TAK1 in vascular smooth muscle cells. Scand. J. Immunol. 2013, 77, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, E.; Egea-Zorrilla, A.; Plaza-Diaz, J.; Aragon-Vela, J.; Munoz-Quezada, S.; Tercedor-Sanchez, L.; Abadia-Molina, F. The gut microbiota and its implication in the development of atherosclerosis and related cardiovascular diseases. Nutrients 2020, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Olas, B. Probiotics, prebiotics and synbiotics-A promising strategy in prevention and treatment of cardiovascular diseases? Int. J. Mol. Sci. 2020, 21, 9737. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Sun, L.; Yu, Y.; Fan, H.; Li, X.; Zhang, X.; Zhuo, X.; Guo, M.; Liu, J.; Liu, P.; et al. A gut feeling of statin. Gut Microbes 2024, 16, 2415487. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef]
- Deehan, E.C.; Al Antwan, S.; Witwer, R.S.; Guerra, P.; John, T.; Monheit, L. Revisiting the concepts of prebiotic and prebiotic effect in light of scientific and regulatory progress-A consensus paper from the Global Prebiotic Association. Adv. Nutr. 2024, 15, 100329. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Prebiotics and probiotics in digestive health. Clin. Gastroenterol. Hepatol. 2019, 17, 333–344. [Google Scholar] [CrossRef]
- Oniszczuk, A.; Oniszczuk, T.; Gancarz, M.; Szymanska, J. Role of gut microbiota, probiotics and prebiotics in the cardiovascular diseases. Molecules 2021, 26, 1172. [Google Scholar] [CrossRef]
- Wu, H.; Chiou, J. Potential benefits of probiotics and prebiotics for coronary heart disease and stroke. Nutrients 2021, 13, 2878. [Google Scholar] [CrossRef]
- Nicolucci, A.C.; Hume, M.P.; Martinez, I.; Mayengbam, S.; Walter, J.; Reimer, R.A. Prebiotics reduce body fat and alter intestinal microbiota in children who are overweight or with obesity. Gastroenterology 2017, 153, 711–722. [Google Scholar] [CrossRef]
- Vulevic, J.; Juric, A.; Tzortzis, G.; Gibson, G.R. A mixture of trans-galactooligosaccharides reduces markers of metabolic syndrome and modulates the fecal microbiota and immune function of overweight adults. J. Nutr. 2013, 143, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, Q.; Huangfu, N.; Cui, H. The effect and mechanism of inulin on atherosclerosis is mediated by the characteristic intestinal flora and metabolites. Coron. Artery Dis. 2024, 35, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Diaz, J.; Ruiz-Ojeda, F.J.; Gil-Campos, M.; Gil, A. Mechanisms of action of probiotics. Adv. Nutr. 2019, 10, S49–S66. [Google Scholar] [CrossRef] [PubMed]
- Latif, A.; Shehzad, A.; Niazi, S.; Zahid, A.; Ashraf, W.; Iqbal, M.W.; Rehman, A.; Riaz, T.; Aadil, R.M.; Khan, I.M.; et al. Probiotics: Mechanism of action, health benefits and their application in food industries. Front. Microbiol. 2023, 14, 1216674. [Google Scholar] [CrossRef] [PubMed]
- Phumkhachorn, P.; Rattanachaikunsopon, P. Probiotics: Sources, selection and health benefits. Res. J. Biotechnol. 2023, 18, 102–113. [Google Scholar] [CrossRef]
- Din, A.U.; Hassan, A.; Zhu, Y.; Yin, T.; Gregersen, H.; Wang, G. Amelioration of TMAO through probiotics and its potential role in atherosclerosis. Appl. Microbiol. Biotechnol. 2019, 103, 9217–9228. [Google Scholar] [CrossRef]
- Reis, S.A.; Conceicao, L.L.; Rosa, D.D.; Siqueira, N.P.; Peluzio, M.C.G. Mechanisms responsible for the hypocholesterolaemic effect of regular consumption of probiotics. Nutr. Res. Rev. 2017, 30, 36–49. [Google Scholar] [CrossRef]
- Prete, R.; Long, S.L.; Gallardo, A.L.; Gahan, C.G.; Corsetti, A.; Joyce, S.A. Beneficial bile acid metabolism from Lactobacillus plantarum of food origin. Sci. Rep. 2020, 10, 1165. [Google Scholar] [CrossRef]
- Hou, G.; Peng, W.; Wei, L.; Li, R.; Yuan, Y.; Huang, X.; Yin, Y. Lactobacillus delbrueckii interfere with bile acid enterohepatic circulation to regulate cholesterol metabolism of growing-finishing pigs via its bile salt hydrolase activity. Front. Nutr. 2020, 7, 617676. [Google Scholar] [CrossRef]
- Tsai, C.C.; Lin, P.P.; Hsieh, Y.M.; Zhang, Z.Y.; Wu, H.C.; Huang, C.C. Cholesterol-lowering potentials of lactic acid bacteria based on bile-salt hydrolase activity and effect of potent strains on cholesterol metabolism In Vitro and In Vivo. Sci. World J. 2014, 2014, 690752. [Google Scholar] [CrossRef]
- El Hage, R.; Al-Arawe, N.; Hinterseher, I. The role of the gut microbiome and trimethylamine oxide in atherosclerosis and age-related disease. Int. J. Mol. Sci. 2023, 24, 2399. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Tao, X.; Xiong, H.; Yu, J.; Wei, H. Lactobacillus plantarum ZDY04 exhibits a strain-specific property of lowering TMAO via the modulation of gut microbiota in mice. Food Funct. 2018, 9, 4299–4309. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, W.; Li, Y.; Luo, S.; Liu, Q.; Zhong, Y.; Jian, Z.; Bao, M. Lactobacillus acidophilus ATCC 4356 attenuates the atherosclerotic progression through modulation of oxidative stress and inflammatory process. Int. Immunopharmacol. 2013, 17, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Khongrum, J.; Yingthongchai, P.; Boonyapranai, K.; Wongtanasarasin, W.; Aobchecy, P.; Tateing, S.; Prachansuwan, A.; Sitdhipol, J.; Niwasabutra, K.; Thaveethaptaikul, P.; et al. Safety and effects of Lactobacillus paracasei TISTR 2593 supplementation on improving cholesterol metabolism and atherosclerosis-related parameters in subjects with hypercholesterolemia: A randomized, double-blind, placebo-controlled clinical trial. Nutrients 2023, 15, 661. [Google Scholar] [CrossRef]
- Keleszade, E.; Kolida, S.; Costabile, A. The cholesterol lowering efficacy of Lactobacillus plantarum ECGC 13110402 in hypercholesterolemic adults: A double-blind, randomized, placebo controlled, pilot human intervention study. J. Funct. Foods 2022, 89, 104939. [Google Scholar] [CrossRef]
- Tenorio-Jimenez, C.; Martinez-Ramirez, M.J.; Del Castillo-Codes, I.; Arraiza-Irigoyen, C.; Tercero-Lozano, M.; Camacho, J.; Chueca, N.; Garcia, F.; Olza, J.; Plaza-Diaz, J.; et al. Lactobacillus reuteri V3401 reduces inflammatory biomarkers and modifies the gastrointestinal microbiome in adults with metabolic syndrome: The PROSIR study. Nutrients 2019, 11, 1761. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Parent, M.; Prakash, S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase-active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012, 107, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Probiotics for Vacular Iflammation in Mtabolic Sndrome (PROMISE)—Clinical Trial NCT03719794. Available online: https://clinicaltrials.gov/study/NCT03719794 (accessed on 1 June 2025).
- Cha, R.R.; Sonu, I. Fecal microbiota transplantation: Present and future. Clin. Endosc. 2025, 58, 352–359. [Google Scholar] [CrossRef]
- Chen, J.H.; Chiu, C.H.; Chen, C.C.; Chen, Y.C.; Yeh, P.J.; Kuo, C.J.; Chiu, C.T.; Cheng, H.T.; Pan, Y.B.; Le, P.H. Comparative efficacy of fecal microbiota transplantation in treating refractory or recurrent Clostridioides difficile infection among patients with and without inflammatory bowel disease: A retrospective cohort study. Biomedicines 2024, 12, 1369. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kassam, Z.; Mullish, B.H.; Chiang, A.; Carrellas, M.; Hurtado, J.; Marchesi, J.R.; McDonald, J.A.K.; Pechlivanis, A.; Barker, G.F.; et al. Effects of fecal microbiota transplantation with oral capsules in obese patients. Clin. Gastroenterol. Hepatol. 2020, 18, 855–863.e2. [Google Scholar] [CrossRef]
- Leshem, A.; Horesh, N.; Elinav, E. Fecal microbial transplantation and its potential application in cardiometabolic syndrome. Front. Immunol. 2019, 10, 1341. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Laurans, L.; Mouttoulingam, N.; Chajadine, M.; Lavelle, A.; Diedisheim, M.; Bacquer, E.; Creusot, L.; Suffee, N.; Esposito, B.; Melhem, N.J.; et al. An obesogenic diet increases atherosclerosis through promoting microbiota dysbiosis-induced gut lymphocyte trafficking into the periphery. Cell Rep. 2023, 42, 113350. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Yoon, B.H.; Lee, S.M.; Choi, M.; Kim, E.H.; Lee, B.W.; Kim, S.Y.; Pack, C.G.; Sung, Y.H.; Baek, I.J.; et al. Fecal microbiota transplantation ameliorates atherosclerosis in mice with C1q/TNF-related protein 9 genetic deficiency. Exp. Mol. Med. 2022, 54, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Metabolic and Metagenomic Effects of Intestinal Microbiome Repopulation in Unexplained Atherosclerosis—Clinical Trial NCT04410003. Available online: https://clinicaltrials.gov/study/NCT04410003 (accessed on 1 June 2025).
- da Ponte Neto, A.M.; Clemente, A.C.O.; Rosa, P.W.; Ribeiro, I.B.; Funari, M.P.; Nunes, G.C.; Moreira, L.; Sparvoli, L.G.; Cortez, R.; Taddei, C.R.; et al. Fecal microbiota transplantation in patients with metabolic syndrome and obesity: A randomized controlled trial. World J. Clin. Cases 2023, 11, 4612–4624. [Google Scholar] [CrossRef]
- Smits, L.P.; Kootte, R.S.; Levin, E.; Prodan, A.; Fuentes, S.; Zoetendal, E.G.; Wang, Z.; Levison, B.S.; Cleophas, M.C.P.; Kemper, E.M.; et al. Effect of vegan fecal microbiota transplantation on carnitine- and choline-derived trimethylamine-N-oxide production and vascular inflammation in patients with metabolic syndrome. J. Am. Heart Assoc. 2018, 7, e008342. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, T.; Costa, V.; Nunes, C.; da Silva, G.J.; Domingues, S. From Dysbiosis to Cardiovascular Disease: The Impact of Gut Microbiota on Atherosclerosis and Emerging Therapies. Appl. Sci. 2025, 15, 7084. https://doi.org/10.3390/app15137084
Lima T, Costa V, Nunes C, da Silva GJ, Domingues S. From Dysbiosis to Cardiovascular Disease: The Impact of Gut Microbiota on Atherosclerosis and Emerging Therapies. Applied Sciences. 2025; 15(13):7084. https://doi.org/10.3390/app15137084
Chicago/Turabian StyleLima, Tiago, Verónica Costa, Carla Nunes, Gabriela Jorge da Silva, and Sara Domingues. 2025. "From Dysbiosis to Cardiovascular Disease: The Impact of Gut Microbiota on Atherosclerosis and Emerging Therapies" Applied Sciences 15, no. 13: 7084. https://doi.org/10.3390/app15137084
APA StyleLima, T., Costa, V., Nunes, C., da Silva, G. J., & Domingues, S. (2025). From Dysbiosis to Cardiovascular Disease: The Impact of Gut Microbiota on Atherosclerosis and Emerging Therapies. Applied Sciences, 15(13), 7084. https://doi.org/10.3390/app15137084