
Synthetic Data Generation for the Development of 2D Gel Electrophoresis Protein Spot Models


Abstract
1. Introduction
- Proposed a method to generate a dataset consisting of synthetic protein spot image samples of high fidelity;
- Developed a new spot modeling algorithm based on autoencoders, using purely the created synthetic dataset;
- Performed a comparative evaluation of the developed protein spot modeling algorithm and other commonly used spot models using synthetic and real data;
- Experimentally showed that the created synthetic dataset is effective and sufficient for the development of protein spot models;
- Publicly provided the synthetic spot dataset generated according to the presented method.
2. Materials and Methods
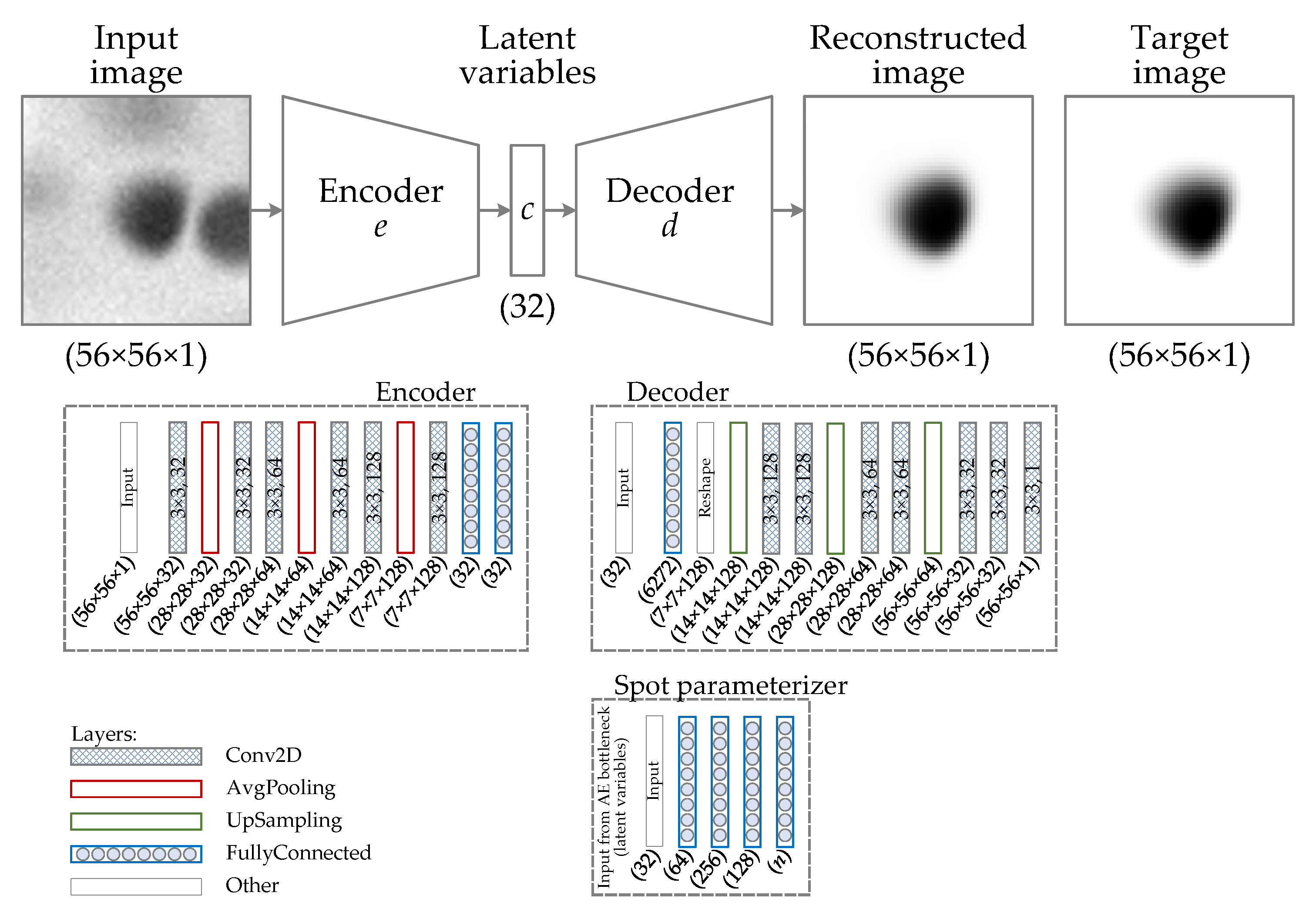
2.1. Generation of Synthetic Protein Spot Images
2.1.1. Requirements for the Synthetic Protein Spot Data
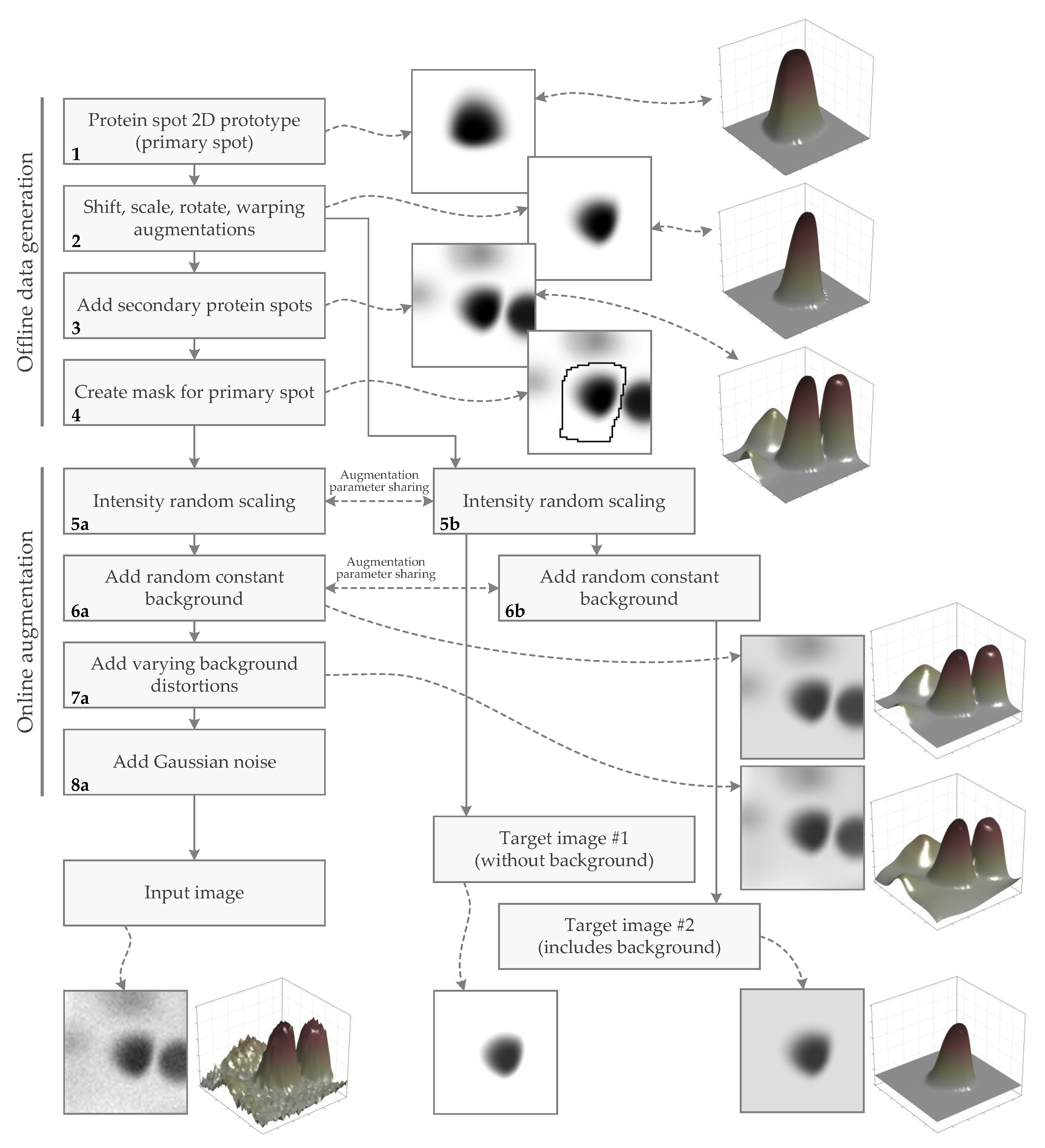
2.1.2. Method for Generating the Synthetic Spot Image Dataset
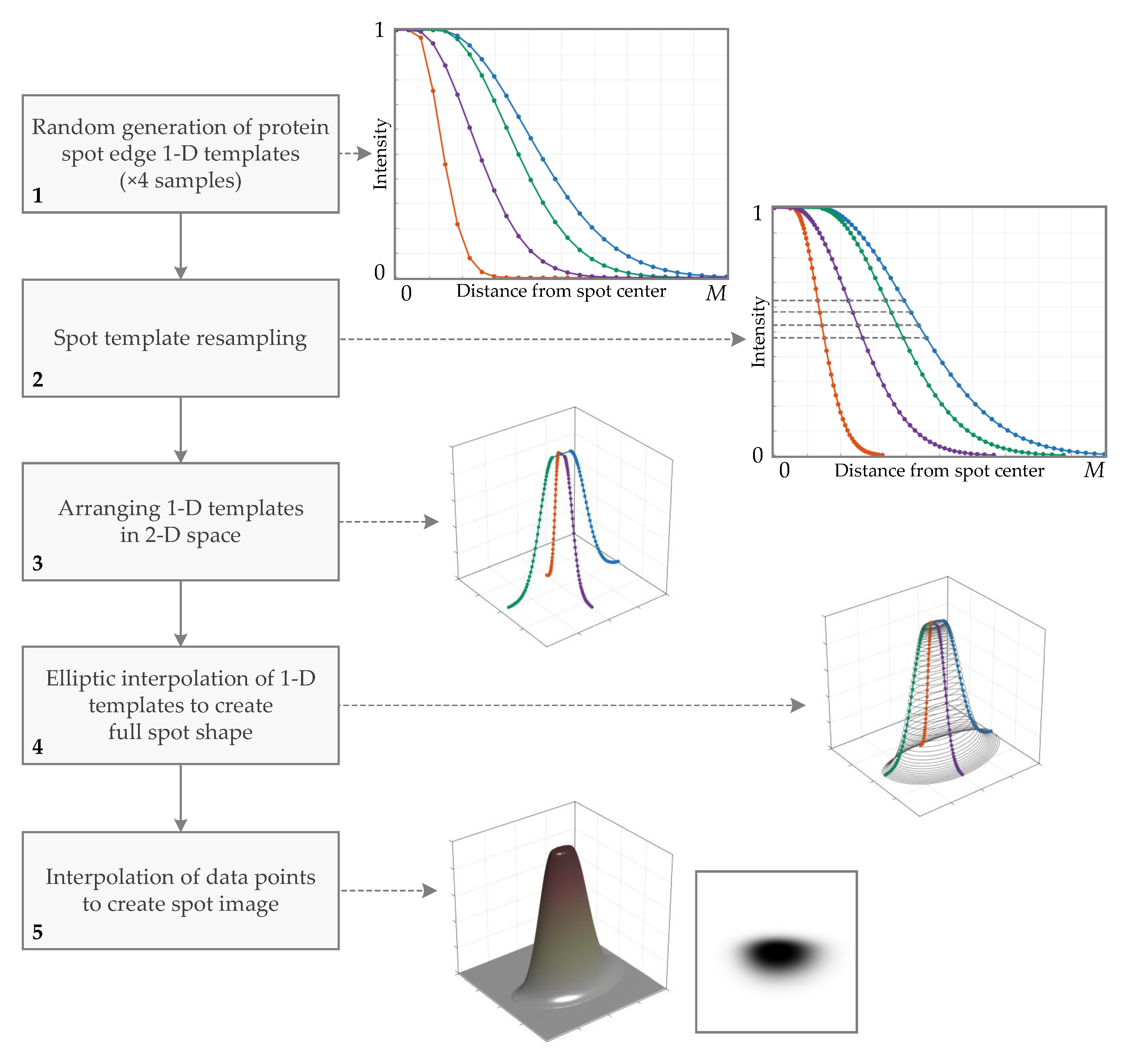
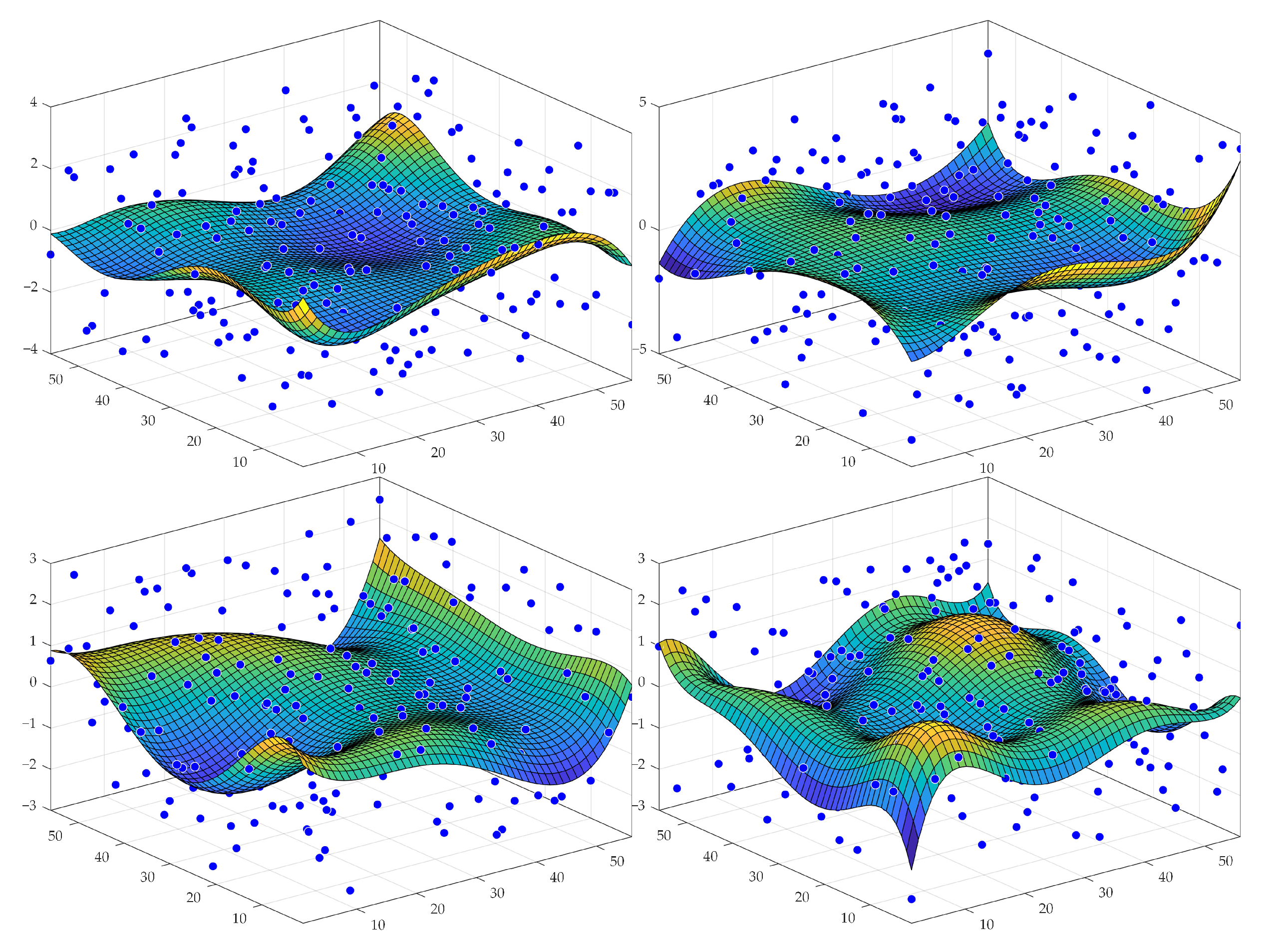
2.1.3. Generation of Protein Spot 2D Prototype (Primary Spot)
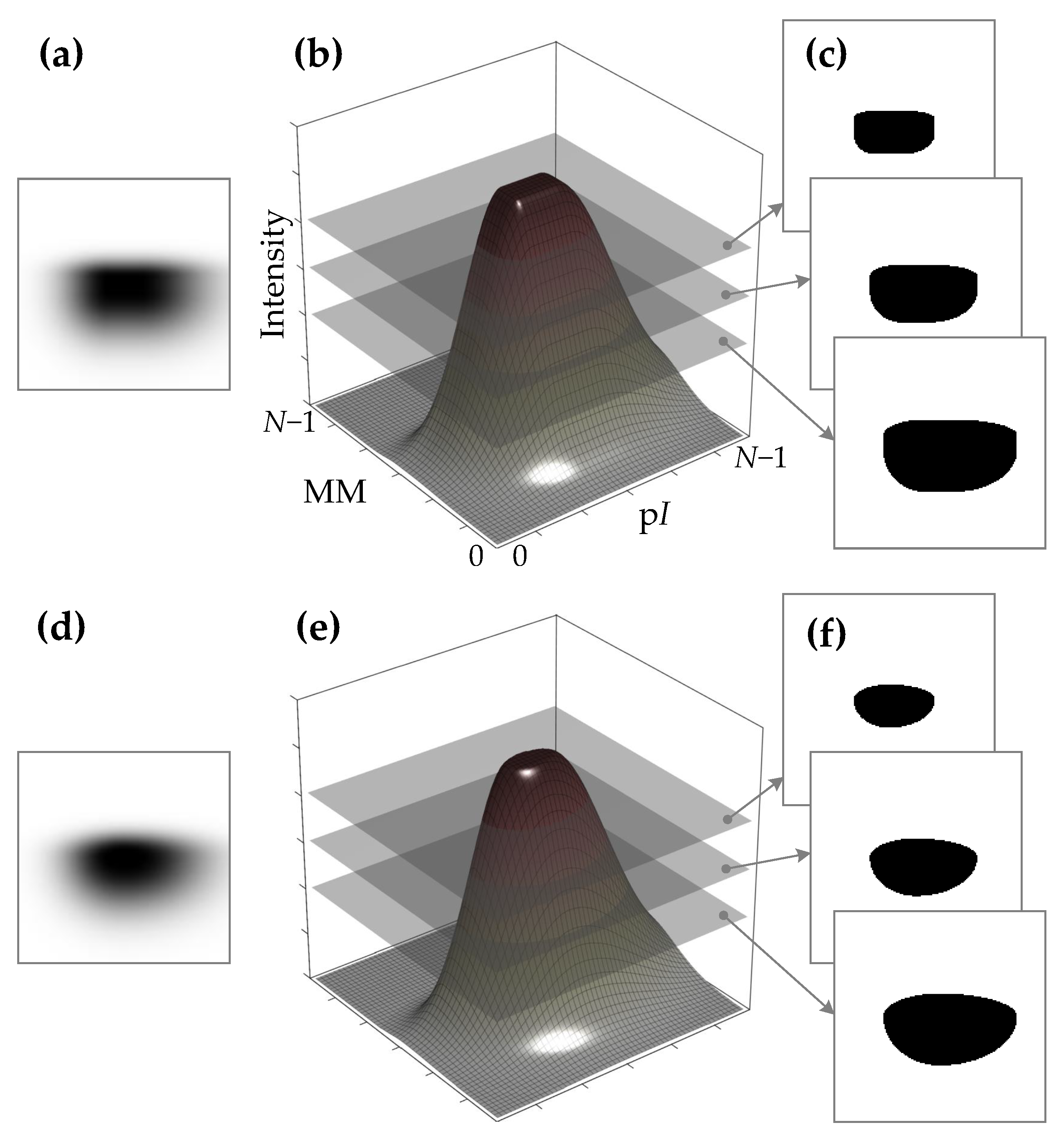
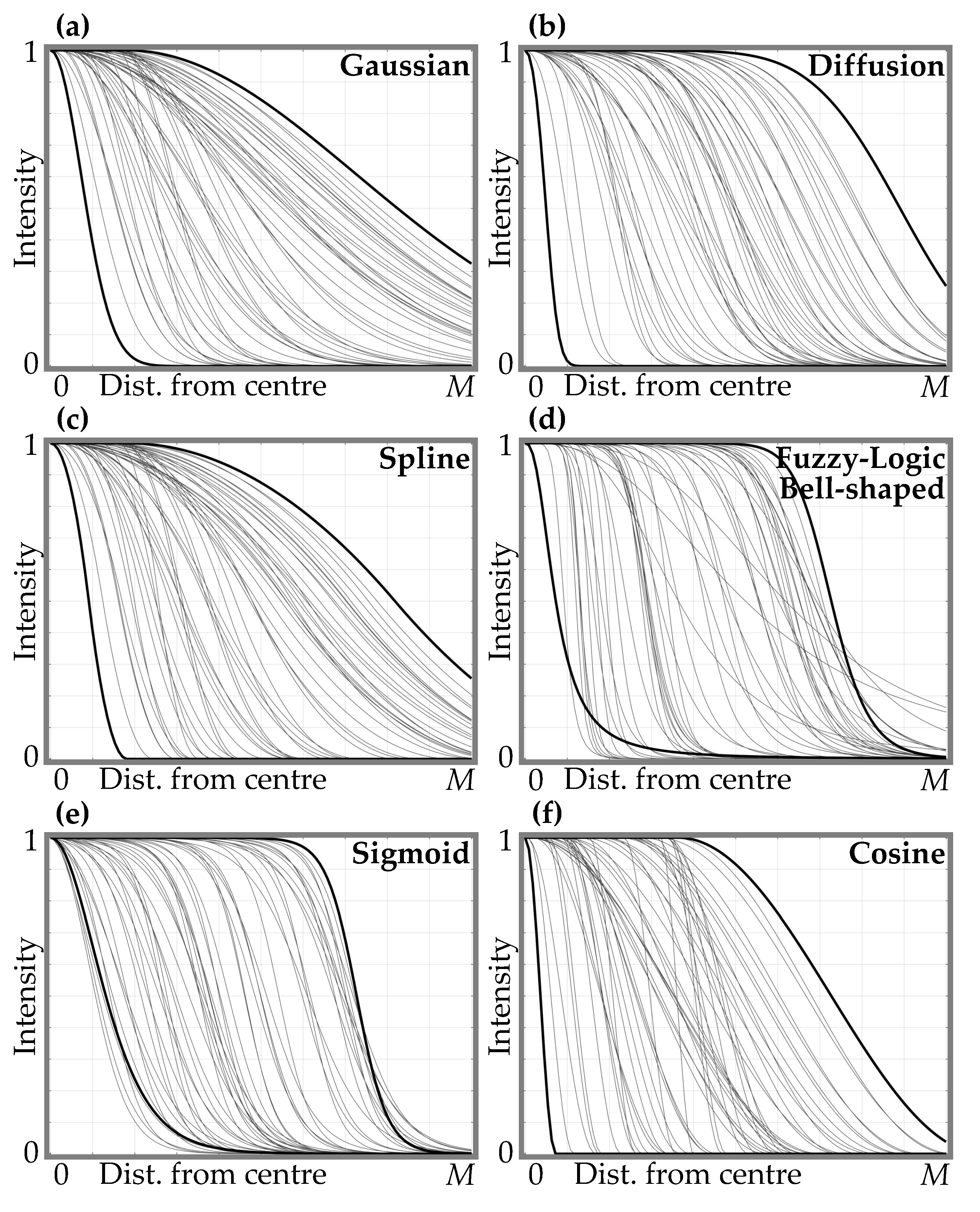
Protein Spot Edge Templates
- Gaussian function with its center at (the graph of the function is shown in Figure 6a):here d —shift of the mean of the Gaussian function from the spot center (); —standard deviation of the Gaussian function.
- Simplified diffusion process function with its center at (refer to Figure 6b):here ; D—parameter defining the diffusion (defines a slope and a shift from the center of the curve transition area); a is the radius of diffusion area.
- Spline function with its center at (refer to Figure 6c):here a is the distance from the center to the beginning of the curve transition area; b is the distance from the a to the end of the curve transition area.
- Fuzzy Logic generalized membership bell-shaped function with its center at (refer to Figure 6d):here a determines the width of the function’s transitional area; b defines the shape of the curve (slope) where a larger value gives a steeper function.
- Sigmoid function with its center at (Figure 6e):here a defines the slope of the transition area of the curve; d—distance from the center to the curve transition area.
- Raised cosine function with its center at (Figure 6f):here a is the width of the transition area of the curve; d—distance from the center to the curve transition area.
Function Parameter Ranges for Edge Template Generation
2.2. Synthetic Data Evaluation
2.3. Software Used
- MATLAB programming and numeric computing platform (version R2021a, The Mathworks Inc., Natick, MA, USA) for the implementation of mathematical protein spot models, for offline data generation of synthetic dataset creation workflow, and for data analysis and visualization;
- Python (version 3.9.10) (https://www.python.org), (accessed on 1 February 2022) [54], an interpreted, high-level, general-purpose programming language. Used for the synthetic dataset creation workflow and for machine learning applications;
- TensorFlow with Keras (version 2.6.0) (https://www.tensorflow.org, (accessed on 1 February 2022)) [55], an open-source platform for machine learning. Used for the online data augmentation stage of the synthetic dataset creation workflow and for the training of autoencoders;
- Albumentations (version 1.0.3) (https://albumentations.ai, (accessed on 1 February 2022)) [50], a Python library for fast and flexible image augmentation. Used for the synthetic dataset creation workflow;
- OpenCV (version 4.5.1) (https://opencv.org/, (accessed on 1 February 2022)) [56], an open source computer vision library. Used for image input/output and manipulations.
2.4. Data Collection
- Synthetic data: the created synthetic test set of spot images is fully and openly available. It can be found at Mendeley Data (https://doi.org/10.17632/x62kt53nnr.1), (accessed on 25 April 2022) The test set consists of 40,000 samples. The training set is not shared online due to its size, but it is similar to the test set as both sets were generated using the same method.
- Real test data: a test set of spots is compiled from real 2DGE images. Spots in images are detected and segmented using the method described in [46,57]. The minimum size of the cropped image patches is px. Crop boxes are centered at the spot centers. The crop box must fully contain the region of the spot that is defined by the mask image. If necessary, the crop box is expanded to fully include the spot region. The test set consists of 6725 samples. 2DGE images were obtained from the following databases or other types of sources:
- -
- Proteome 2D-PAGE Database (https://protein.mpiib-berlin.mpg.de/cgi-bin/pdbs/2d-page/extern/index.cgi), (accessed on 21 February 2022) [58];
- -
- The LECB 2-D PAGE Gel Images Data Sets (http://bioinformatics.org/lecb2dgeldb/), (accessed on 21 February 2022);
- -
- The Human Myocardial Two-Dimensional Electrophoresis Protein Database (HEART-2DPAGE) (http://www.chemie.fu-berlin.de/user/pleiss/), (accessed on 21 February 2022) [59];
- -
- HeLa Study Reference Images (http://www.fixingproteomics.org/advice/helastandard.html), (accessed on 11 August 2010) [60,61];
- -
- Images used in the validation of RAIN algorithm [62] (http://www.proteomegrid.org/rain/, (accessed on 20 July 2009)).
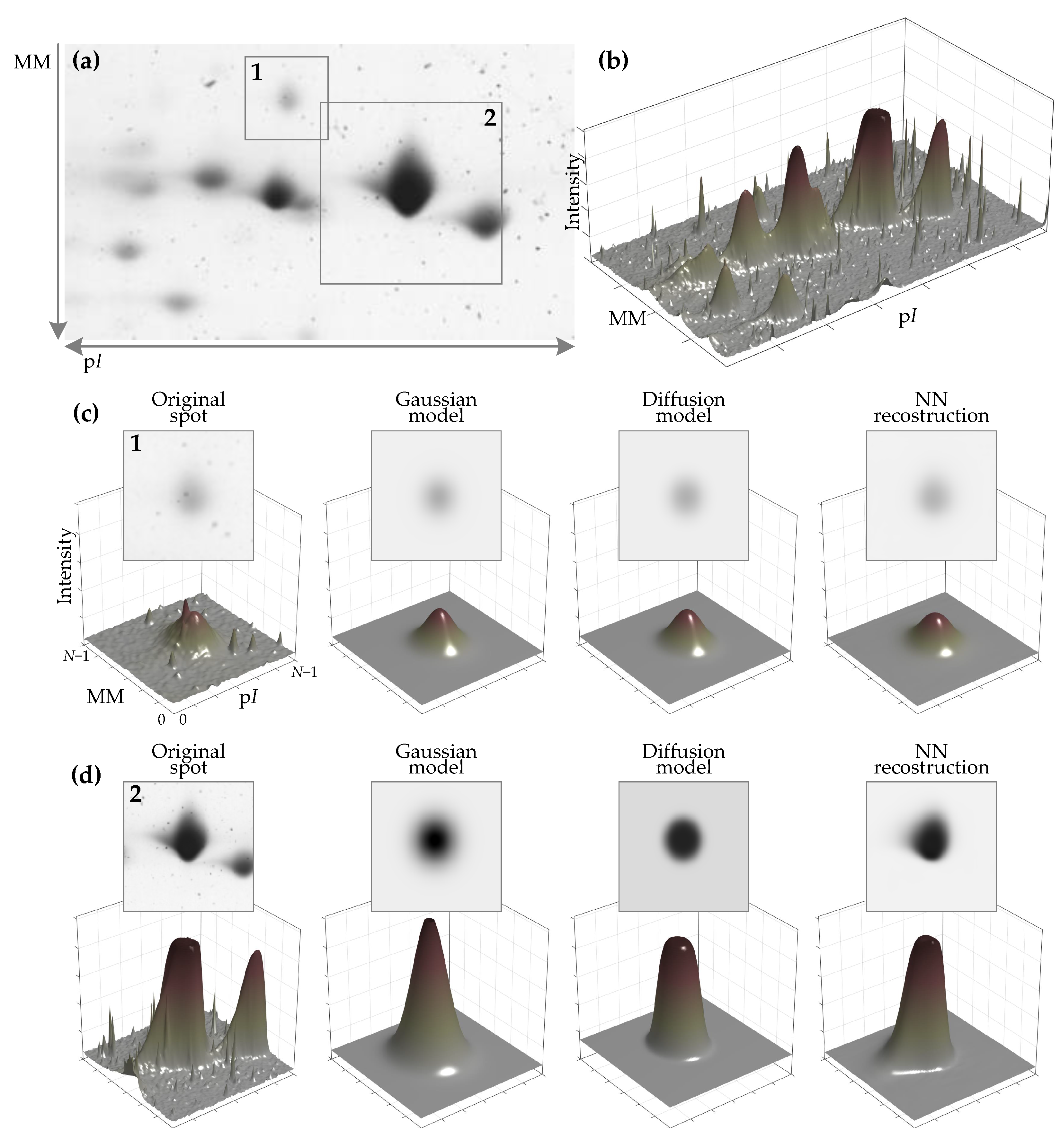
3. Results and Discussion
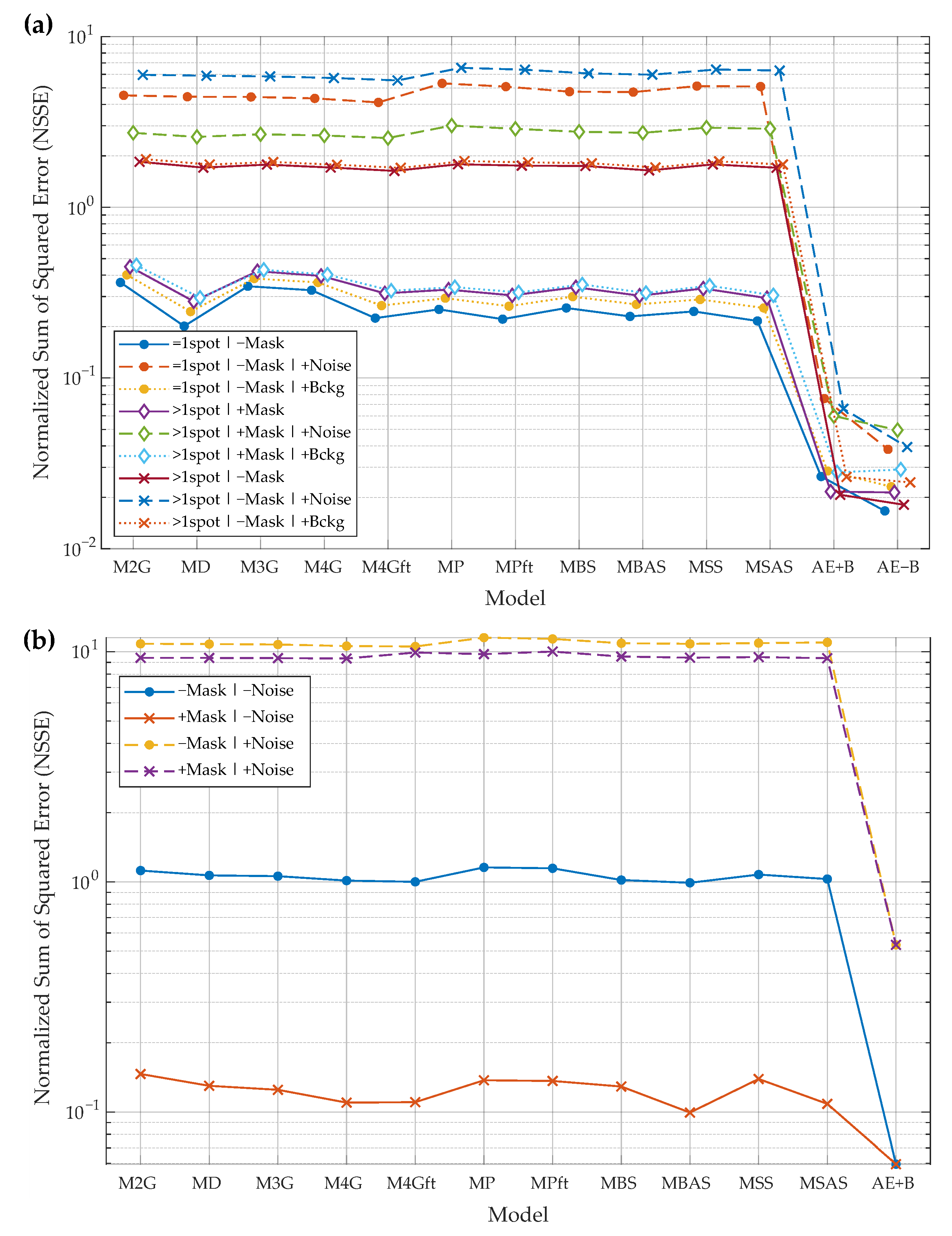
- Reconstruction of synthetic images:
- -
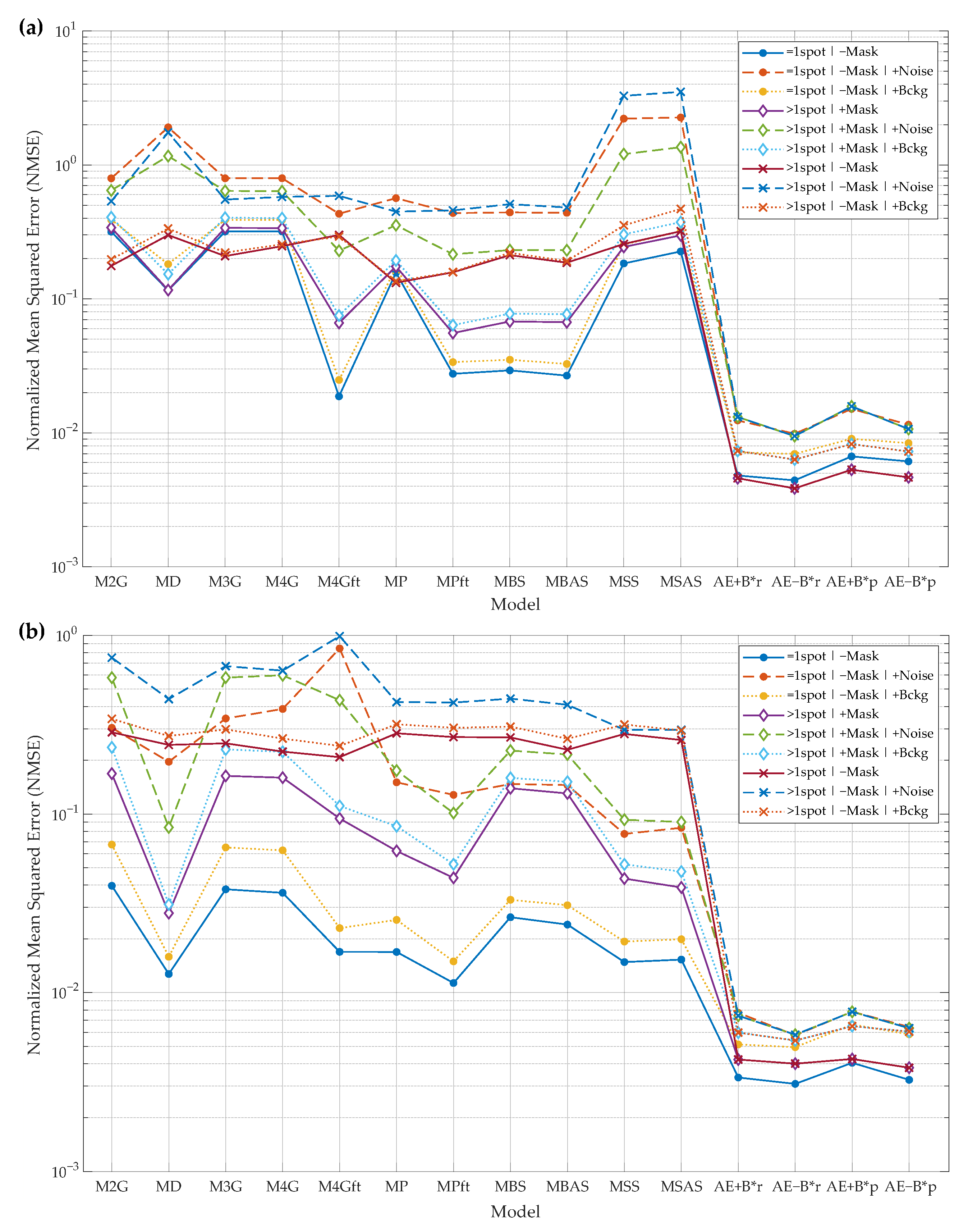
- input image may contain only the primary spot or the primary spot surrounded by secondary spots. These cases are marked by “=1spot” or “>1spot”, respectively, in the graphs of Figure 8a;
- -
- during the model fitting and the evaluation of reconstruction residual error, the mask of the primary spot may or may not be used. The baseline algorithms use a mask to fit a model only in the mask area. AE does not use the mask as additional input data. All algorithms use the mask for residual error evaluation if the mask is provided in a particular experiment. These cases are marked by “+Mask” or “−Mask”, respectively, in the graphs of Figure 8a;
- -
- input images may be corrupted by Gaussian noise (marked by “+Noise”), by slowly varying background (marked by “+Bckg”), or neither (no additional marking);
- -
- two AE models are trained and evaluated—”AE−B” is the model that reconstructs the image with background eliminated and “AE−B” reconstructs the image with the constant background.
- Reconstruction of real images:
- -
- due to the fact that in real 2DGE images the ground-truth spots are unknown, the experimental setup is slightly different. The ground-truth images are simulated, in one experimental case, by smoothing original spot images using a median filter (marked by “−Noise” in Figure 8b). In the other case, the ground-truth images are the same, but the input images are generated by adding Gaussian and Salt & Pepper noise to the ground-truth images (noted by “+Noise” in the legend);
- -
- during the fitting of the baseline models, the mask of the main spot may or may not be used. The mask is always used when evaluating residual errors. Cases are marked by “+Mask” or “−Mask”, respectively, in the graphs of Figure 8b;
- -
- the true background intensity is difficult to estimate, so the AE was used to reconstruct spots with the background (“AE+B” algorithm).
- Two-way adapting (anisotropic) 2D Gaussian model (M2G);
- The diffusion model (MD);
- Three Gaussian functions model (M3G);
- Four Gaussian functions model (M4G);
- Four Gaussian functions with flat top model (M4GFT);
- Simple -shaped model (MP);
- -shape model with flat top (MPft);
- Two-way symmetric bell-shaped model (MBS);
- Asymmetric bell-shaped model (MBAS);
- Two-way symmetric sigmoid-based model (MSS);
- Asymmetric sigmoid-based model (MSAS).
- “AE+B”—spot reconstructed by the autoencoder with the constant background remaining;
- “AE−B”—spot reconstructed by the autoencoder without background;
- “AE+B*r”—spot parameters computed from the reconstructed spot, and background is not removed during reconstruction;
- “AE−B*r”—spot parameters computed from the reconstructed spot, and background is removed during reconstruction;
- “AE+B*p”—spot parameters predicted from the AE’s bottleneck features, and AE is trained to reconstruct the spot leaving the background;
- “AE−B*p”—spot parameters predicted from the AE’s bottleneck features, and AE is trained to reconstruct the spot without background.
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2DGE, 2DE | Two-dimensional Gel Electrophoresis |
| ML | Machine Learning |
| AI | Artificial Intelligence |
| AE | Autoencoder |
| ANN | Artificial Neural Network |
| CNN | Convolutional Neural Network |
| LR | Learning Rate |
| MSE | Mean Squared Error |
| NMSE | Normalized Mean Squared Error |
| SSE | Sum of Squared Error |
| NSSE | Normalized Sum of Squared Error |
References
- O’Farrell, P. High Resolution 2-Dimensional Electrophoresis of Proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Moche, M.; Albrecht, D.; Maaß, S.; Hecker, M.; Westermeier, R.; Büttner, K. The new horizon in 2D electrophoresis: New technology to increase resolution and sensitivity. Electrophoresis 2013, 34, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.N.; Seok, S.J.; Kim, H.K.; Kim, G.H.; Yang, J.O. Comparative Proteomics Analysis of Phosphine-Resistant and Phosphine-Susceptible Sitophilus oryzae (Coleoptera: Curculionidae). Appl. Sci. 2021, 11, 4163. [Google Scholar] [CrossRef]
- Venugopal, D.C.; Ravindran, S.; Shyamsundar, V.; Sankarapandian, S.; Krishnamurthy, A.; Sivagnanam, A.; Madhavan, Y.; Ramshankar, V. Integrated Proteomics Based on 2D Gel Electrophoresis and Mass Spectrometry with Validations: Identification of a Biomarker Compendium for Oral Submucous Fibrosis—An Indian Study. J. Pers. Med. 2022, 12, 208. [Google Scholar] [CrossRef]
- Guzmán-Flores, J.M.; Flores-Pérez, E.C.; Hernández-Ortiz, M.; Vargas-Ortiz, K.; Ramírez-Emiliano, J.; Encarnación-Guevara, S.; Pérez-Vázquez, V. Protein expression profile of twenty-week-old diabetic db/db and non-diabetic mice livers: A proteomic and bioinformatic analysis. Biomolecules 2018, 8, 35. [Google Scholar] [CrossRef]
- Ura, B.; Biffi, S.; Monasta, L.; Arrigoni, G.; Battisti, I.; Di Lorenzo, G.; Romano, F.; Aloisio, M.; Celsi, F.; Addobbati, R.; et al. Two Dimensional-Difference in Gel Electrophoresis (2D-DIGE) Proteomic Approach for the Identification of Biomarkers in Endometrial Cancer Serum. Cancers 2021, 13, 3639. [Google Scholar] [CrossRef]
- Rogowska-Wrzesinska, A.; Le Bihan, M.C.; Thaysen-Andersen, M.; Roepstorff, P. 2D gels still have a niche in proteomics. J. Proteom. 2013, 88, 4–13. [Google Scholar] [CrossRef]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de Souza, D. 2DE: The Phoenix of Proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef]
- Abdallah, C.; Dumas-Gaudot, E.; Renaut, J.; Sergeant, K. Gel-based and gel-free quantitative proteomics approaches at a glance. Int. J. Plant Genom. 2012, 2012. [Google Scholar] [CrossRef]
- Kim, Y.I.; Cho, J.Y. Gel-based proteomics in disease research: Is it still valuable? Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 9–16. [Google Scholar] [CrossRef]
- Bocian, A.; Buczkowicz, J.; Jaromin, M.; Hus, K.K.; Legáth, J. An effective method of isolating honey proteins. Molecules 2019, 24, 2399. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Chevallet, M.; Luche, S.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: Past, present and future. J. Proteom. 2010, 73, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Saraygord-Afshari, N.; Low, T.Y. The evolution of two-dimensional gel electrophoresis-from proteomics to emerging alternative applications. J. Chromatogr. A 2020, 1615, 460763. [Google Scholar] [CrossRef] [PubMed]
- Fulton, K.M.; Twine, S.M. Immunoproteomics: Current technology and applications. In Immunoproteomics; Springer: Berlin/Heidelberg, Germany, 2013; pp. 21–57. [Google Scholar]
- Leber, T.M.; Balkwill, F.R. Zymography: A single-step staining method for quantitation of proteolytic activity on substrate gels. Anal. Biochem. 1997, 249, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Jayathilaka, L.P.; Huang, J.S.; Gupta, S. Applications of immobilized metal affinity electrophoresis. In Electrophoretic Separation of Proteins; Springer: Berlin/Heidelberg, Germany, 2019; pp. 371–385. [Google Scholar]
- Werhahn, W.; Braun, H.P. Biochemical dissection of the mitochondrial proteome from Arabidopsis thaliana by three-dimensional gel electrophoresis. Electrophoresis 2002, 23, 640–646. [Google Scholar] [CrossRef]
- Valledor, L.; Jorrín, J. Back to the basics: Maximizing the information obtained by quantitative two dimensional gel electrophoresis analyses by an appropriate experimental design and statistical analyses. J. Proteom. 2011, 74, 1–18. [Google Scholar] [CrossRef]
- Schnaars, V.; Dörries, M.; Hutchins, M.; Wöhlbrand, L.; Rabus, R. What’s the Difference? 2D DIGE Image Analysis by DeCyderTM versus SameSpotsTM. J. Mol. Microbiol. Biotechnol. 2018, 28, 128–136. [Google Scholar] [CrossRef]
- Jungblut, P.R. The proteomics quantification dilemma. J. Proteom. 2014, 107, 98–102. [Google Scholar] [CrossRef]
- Brandão, A.; Barbosa, H.; Arruda, M. Image analysis of two-dimensional gel electrophoresis for comparative proteomics of transgenic and non-transgenic soybean seeds. J. Proteom. 2010, 73, 1433–1440. [Google Scholar] [CrossRef]
- Molina-Mora, J.A.; Chinchilla-Montero, D.; Castro-Peña, C.; García, F. Two-dimensional gel electrophoresis (2D-GE) image analysis based on CellProfiler: Pseudomonas aeruginosa AG1 as model. Medicine 2020, 99, e23373. [Google Scholar] [CrossRef]
- Dowsey, A.W.; English, J.A.; Lisacek, F.; Morris, J.S.; Yang, G.Z.; Dunn, M.J. Image analysis tools and emerging algorithms for expression proteomics. Proteomics 2010, 10, 4226–4257. [Google Scholar] [CrossRef] [PubMed]
- Natale, M.; Maresca, B.; Abrescia, P.; Bucci, E. Image analysis workflow for 2-D electrophoresis gels based on ImageJ. Proteom. Insights 2011, 4, 37–49. [Google Scholar] [CrossRef]
- Morris, J.S.; Clark, B.N.; Wei, W.; Gutstein, H.B. Evaluating the performance of new approaches to spot quantification and differential expression in 2-dimensional gel electrophoresis studies. J. Proteome Res. 2009, 9, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Berth, M.; Moser, F.M.; Kolbe, M.; Bernhardt, J. The state of the art in the analysis of two-dimensional gel electrophoresis images. Appl. Microbiol. Biotechnol. 2007, 76, 1223–1243. [Google Scholar] [CrossRef] [PubMed]
- Srinark, T.; Kambhamettu, C. An image analysis suite for spot detection and spot matching in two-dimensional electrophoresis gels. Electrophoresis 2008, 29, 706–715. [Google Scholar] [CrossRef]
- Brauner, J.M.; Groemer, T.W.; Stroebel, A.; Grosse-Holz, S.; Oberstein, T.; Wiltfang, J.; Kornhuber, J.; Maler, J.M. Spot quantification in two dimensional gel electrophoresis image analysis: Comparison of different approaches and presentation of a novel compound fitting algorithm. BMC Bioinform. 2014, 15, 181. [Google Scholar] [CrossRef]
- Li, F.; Seillier-Moiseiwitsch, F.; Korostyshevskiy, V.R. Region-based statistical analysis of 2D PAGE images. Comput. Stat. Data Anal. 2011, 55, 3059–3072. [Google Scholar] [CrossRef]
- Millioni, R.; Puricelli, L.; Sbrignadello, S.; Iori, E.; Murphy, E.; Tessari, P. Operator-and software-related post-experimental variability and source of error in 2-DE analysis. Amino Acids 2012, 42, 1583–1590. [Google Scholar] [CrossRef]
- Kostopoulou, E.; Katsigiannis, S.; Maroulis, D. SpotDSQ: A 2D-gel image analysis tool for protein spot detection, segmentation and quantification. In Proceedings of the 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE), Athens, Greece, 28–30 October 2019; pp. 31–37. [Google Scholar]
- Goez, M.M.; Torres-Madronero, M.C.; Rothlisberger, S.; Delgado-Trejos, E. Joint pre-processing framework for two-dimensional gel electrophoresis images based on nonlinear filtering, background correction and normalization techniques. BMC Bioinform. 2020, 21, 376. [Google Scholar] [CrossRef]
- Sengar, R.S.; Upadhyay, A.K.; Singh, M.; Gadre, V.M. Analysis of 2D-gel images for detection of protein spots using a novel non-separable wavelet based method. Biomed. Signal Process. Control. 2016, 25, 62–75. [Google Scholar] [CrossRef]
- Nhek, S.; Tessema, B.; Indahl, U.; Martens, H.; Mosleth, E. 2D electrophoresis image segmentation within a pixel-based framework. Chemom. Intell. Lab. Syst. 2015, 141, 33–46. [Google Scholar] [CrossRef]
- Shamekhi, S.; Baygi, M.H.M.; Azarian, B.; Gooya, A. A novel multi-scale Hessian based spot enhancement filter for two dimensional gel electrophoresis images. Comput. Biol. Med. 2015, 66, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, E.; Zacharia, E.; Maroulis, D. An Effective Approach for Detection and Segmentation of Protein Spots on 2-D Gel Images. IEEE J. Biomed. Health Inform. 2014, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- dos Anjos, A.; Møller, A.L.; Ersbøll, B.K.; Finnie, C.; Shahbazkia, H.R. New approach for segmentation and quantification of two-dimensional gel electrophoresis images. Bioinformatics 2011, 27, 368–375. [Google Scholar] [CrossRef][Green Version]
- Morris, J.S.; Clark, B.N.; Gutstein, H.B. Pinnacle: A fast, automatic and accurate method for detecting and quantifying protein spots in 2-dimensional gel electrophoresis data. Bioinformatics 2008, 24, 529–536. [Google Scholar] [CrossRef]
- Kostopoulou, E.; Katsigiannis, S.; Maroulis, D. 2D-gel spot detection and segmentation based on modified image-aware grow-cut and regional intensity information. Comput. Methods Programs Biomed. 2015, 122, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lozano, C.; Seoane, J.A.; Gestal, M.; Gaunt, T.R.; Dorado, J.; Pazos, A.; Campbell, C. Texture analysis in gel electrophoresis images using an integrative kernel-based approach. Sci. Rep. 2016, 6, 19256. [Google Scholar] [CrossRef]
- Goez, M.M.; Torres-Madroñero, M.C.; Röthlisberger, S.; Delgado-Trejos, E. Preprocessing of 2-dimensional gel electrophoresis images applied to proteomic analysis: A review. Genom. Proteom. Bioinform. 2018, 16, 63–72. [Google Scholar] [CrossRef]
- Garrels, J.I. The QUEST system for quantitative analysis of two-dimensional gels. J. Biol. Chem. 1989, 264, 5269–5282. [Google Scholar] [CrossRef]
- Marczyk, M. Mixture Modeling of 2-D Gel Electrophoresis Spots Enhances the Performance of Spot Detection. IEEE Trans. Nanobiosci. 2017, 16, 91–99. [Google Scholar] [CrossRef]
- Rogers, M.; Graham, J.; Tonge, P. Using statistical image models for objective evaluation of spot detection in two-dimensional gels. Proteomics 2003, 3, 879–886. [Google Scholar] [CrossRef]
- Bettens, E.; Scheunders, P.; Vandyck, D.; Moens, L.; Vanosta, P. Computer analysis of two-dimensional electrophoresis gels: A new segmentation and modeling algorithm. Electrophoresis 1997, 18, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Navakauskienė, R.; Navakauskas, D.; Borutinskaitė, V.; Matuzevičius, D. Computational Methods for Proteome Analysis. In Epigenetics and Proteomics of Leukemia: A Synergy of Experimental Biology and Computational Informatics; Springer International Publishing: Cham, Switzerland, 2021; pp. 195–282. [Google Scholar] [CrossRef]
- Serackis, A.; Navakauskas, D. Treatment of over-saturated protein spots in two-dimensional electrophoresis gel images. Informatica 2010, 21, 409–424. [Google Scholar] [CrossRef]
- Ahmed, A.S.; El-Behaidy, W.H.; Youssif, A.A. Medical image denoising system based on stacked convolutional autoencoder for enhancing 2-dimensional gel electrophoresis noise reduction. Biomed. Signal Process. Control 2021, 69, 102842. [Google Scholar] [CrossRef]
- NVIDIA. What Is Synthetic Data. Available online: https://blogs.nvidia.com/blog/2021/06/08/what-is-synthetic-data/ (accessed on 14 March 2022).
- Buslaev, A.; Iglovikov, V.I.; Khvedchenya, E.; Parinov, A.; Druzhinin, M.; Kalinin, A.A. Albumentations: Fast and Flexible Image Augmentations. Information 2020, 11, 125. [Google Scholar] [CrossRef]
- Vincent, L.; Soille, P. Watersheds in Digital Spaces—An Efficient Algorithm Based on Immersion Simulations. IEEE Trans. Pattern Anal. Mach. Intell. 1991, 13, 583–598. [Google Scholar] [CrossRef]
- Coleman, T.F.; Li, Y. An interior trust region approach for nonlinear minimization subject to bounds. Siam J. Optim. 1996, 6, 418–445. [Google Scholar] [CrossRef]
- Coleman, T.; Li, Y. On the convergence of reflective newton methods for large-scale nonlinear minimization subject to bounds. Math. Program. 1994, 67, 189–224. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. 2015. Available online: https://www.tensorflow.org (accessed on 1 February 2022).
- Bradski, G. The OpenCV Library. Dr. Dobb’s J. Softw. Tools 2000, 25, 120–123. [Google Scholar]
- Matuzevičius, D.; Žurauskas, E.; Navakauskienė, R.; Navakauskas, D. Improved proteomic characterization of human myocardium and heart conduction system by computational methods. Biologija 2008, 54, 283–289. [Google Scholar] [CrossRef]
- Lange, S.; Rosenkrands, I.; Stein, R.; Andersen, P.; Kaufmann, S.H.; Jungblut, P.R. Analysis of protein species differentiation among mycobacterial low-Mr-secreted proteins by narrow pH range Immobiline gel 2-DE-MALDI-MS. J. Proteom. 2014, 97, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Plei, K.P.; Söding, P.; Sander, S.; Oswald, H.; Neuß, M.; Regitz-Zagrosek, V.; Fleck, E. Dilated cardiomyopathy-associated proteins and their presentation in a WWW-accessible two-dimensional gel protein database. Electrophoresis 1997, 18, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.W.; Deutsch, E.W.; Au, C.E.; Kearney, R.E.; Beavis, R.; Sechi, S.; Nilsson, T.; Bergeron, J.J. A HUPO test sample study reveals common problems in mass spectrometry–based proteomics. Nat. Methods 2009, 6, 423–430. [Google Scholar] [CrossRef]
- Mann, M. Comparative analysis to guide quality improvements in proteomics. Nat. Methods 2009, 6, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Dowsey, A.W.; Dunn, M.J.; Yang, G.Z. Automated image alignment for 2D gel electrophoresis in a high-throughput proteomics pipeline. Bioinformatics 2008, 24, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.; Taylor, J.; Scandora, A.; Coulter, B.; Anderson, N. The TYCHO System For Computer-Analysis of Two-Dimensional Gel-Electrophoresis Patterns. Clin. Chem. 1981, 27, 1807–1820. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter Range | ||||||
|---|---|---|---|---|---|---|
| Function | Parameter #1 | Min | Max | Parameter #2 | Min | Max |
| 1. Gaussian | 2 | 15 | d | 0 | 5 | |
| 2. Diffusion | a | 3.5 | 8 | D | 0.2 | 10 |
| 3. Spline | a | 0 | 5 | b | 5 | 35 |
| 4. FL Bell-shaped | a | 2 | 20 | b | 1.2 | 8 |
| 5. Sigmoid | a | 0.5 | 1 | d | 1 | 20 |
| 6. Cosine | a | 2 | 20 | d | 0 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuzevičius, D. Synthetic Data Generation for the Development of 2D Gel Electrophoresis Protein Spot Models. Appl. Sci. 2022, 12, 4393. https://doi.org/10.3390/app12094393
Matuzevičius D. Synthetic Data Generation for the Development of 2D Gel Electrophoresis Protein Spot Models. Applied Sciences. 2022; 12(9):4393. https://doi.org/10.3390/app12094393
Chicago/Turabian StyleMatuzevičius, Dalius. 2022. "Synthetic Data Generation for the Development of 2D Gel Electrophoresis Protein Spot Models" Applied Sciences 12, no. 9: 4393. https://doi.org/10.3390/app12094393
APA StyleMatuzevičius, D. (2022). Synthetic Data Generation for the Development of 2D Gel Electrophoresis Protein Spot Models. Applied Sciences, 12(9), 4393. https://doi.org/10.3390/app12094393