
iEnhancer-Deep: A Computational Predictor for Enhancer Sites and Their Strength Using Deep Learning




Abstract
:1. Introduction
2. Materials and Methods
2.1. Benchmark Dataset
2.2. Methods
2.3. Gaussian Noise
2.4. Evaluation Parameters
3. Results and Discussion
3.1. Effect of Different Encoding Techniques
3.2. Cross-Validation Test
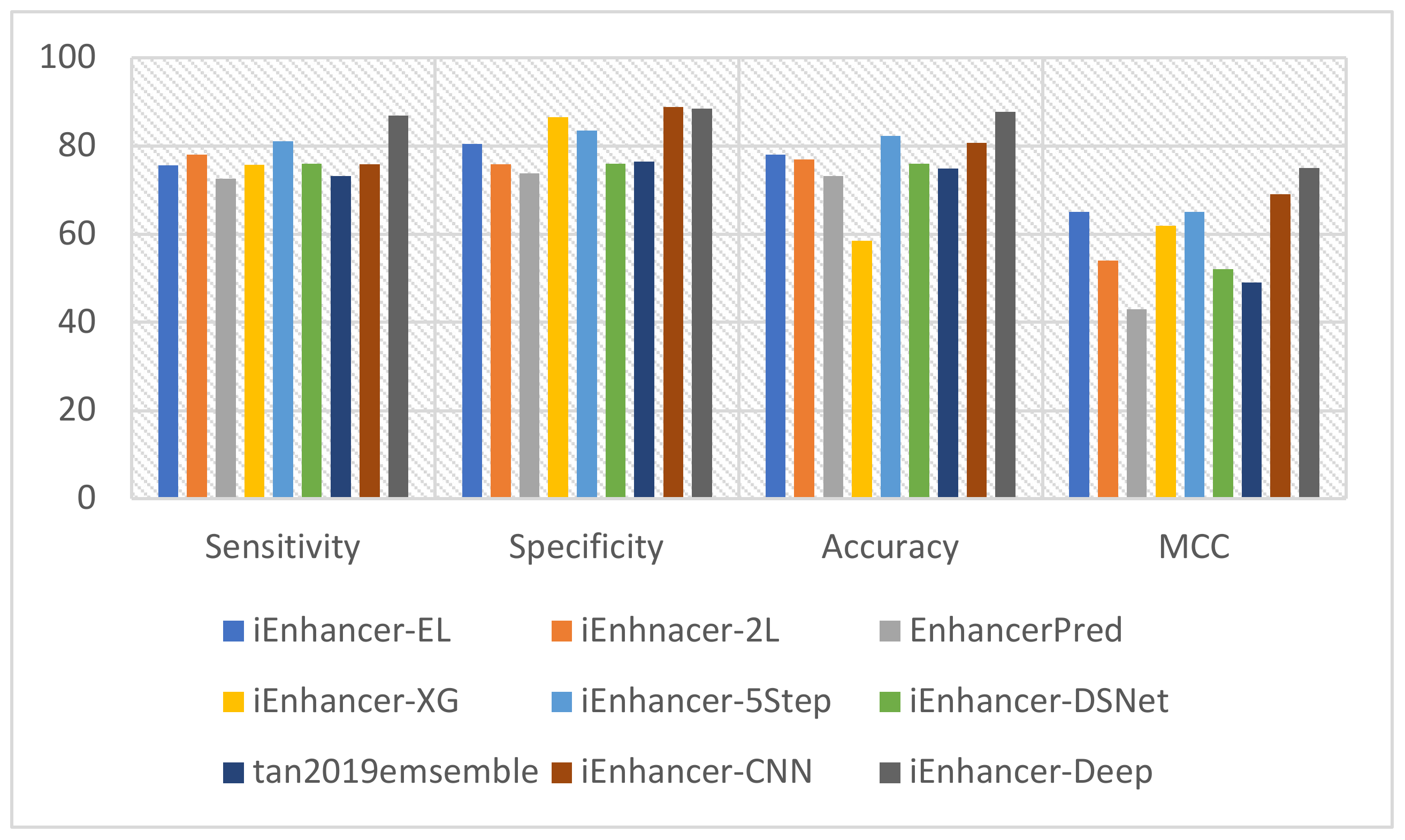
3.3. Performance Comparison with Existing Methods
3.4. Independent Dataset
3.5. Cross-Species Test
4. Web Server
- -
- Click on the following link to access the web server http://nsclbio.jbnu.ac.kr/tools/iEnhancer-Deep/ (accessed on 17 November 2021);
- -
- Options to copy/paste, upload, or simply write the DNA sequence in the input box in FASTS format are available;
- -
- For results, press the ‘submit sequence’ option;
- -
- One thousand is the largest number of sequences that our server can instantaneously sort.
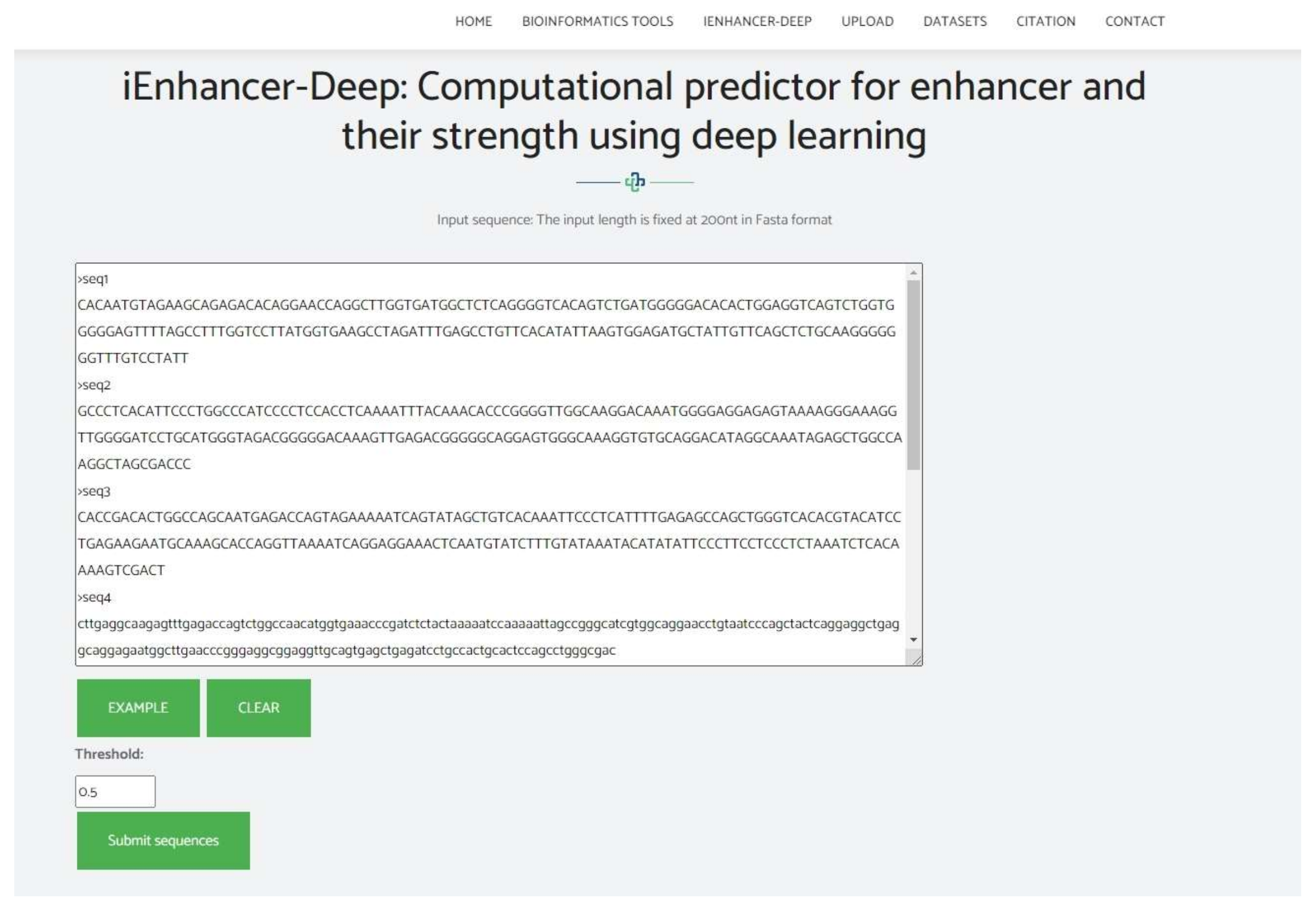
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef]
- Plank, J.L.; Dean, A. Enhancer function: Mechanistic and genomewide insights come together. Mol. Cell 2014, 55, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fang, L.; Long, R.; Lan, X.; Chou, K.-C. Ienhancer-2l: A twolayer predictor for identifying enhancers and their strength by pseudo ktuple nucleotide composition. Bioinformatics 2016, 32, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, M.; Thodberg, M.; Vitezic, M.; Bornholdt, J.; Vitting-Seerup, K.; Chen, Y.; Coskun, M.; Li, Y.; Lo, B.Z.S.; Klausen, P.; et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nat. Commun. 2018, 9, 1–19. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef]
- Firpi, H.A.; Ucar, D.; Tan, K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics 2010, 26, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. Rfecs: A random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef]
- Erwin, G.D.; Oksenberg, N.; Truty, R.M.; Kostka, D.; Murphy, K.K.; Ahituv, N.; Pollard, K.S.; Capra, J.A. Integrating diverse datasets improves developmental enhancer prediction. PLoS Comput. Biol. 2014, 10, e1003677. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.; Miranda-Saavedra, D. Genome-wide enhancer prediction from epigenetic signatures using genetic algorithm-optimized support vector machines. Nucleic Acids Res. 2012, 40, e77. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Lee, D.; Mohammad-Noori, M.; Beer, M.A. Enhanced Regulatory Sequence Prediction Using Gapped k-mer Features. PLoS Comput. Biol. 2014, 10, e1003711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleftogiannis, D.; Kalnis, P.; Bajic, V.B. DEEP: A general computational framework for predicting enhancers. Nucleic Acids Res. 2014, 43, e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, H.; Gan, Y.; Wang, Y.; Zhou, S.; Guan, J. A new method for enhancer prediction based on deep belief network. BMC Bioinform. 2017, 18, 418. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Liu, F.; Ren, C.; Ouyang, Z.; Xie, Z.; Bo, X.; Shu, W. BiRen: Predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 2017, 33, 1930–1936. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, K.; Huang, D.-S.; Chou, K.-C. ienhancer-el: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Khanal, J.; Nazari, I.; Tayara, H.; Chong, K.T. 4mccnn: Identification of n4-methylcytosine sites in prokaryotes using convolutional neural network. IEEE Access 2019, 7, 145455–145461. [Google Scholar] [CrossRef]
- Tayara, H.; Chong, K.T. Improving the Quantification of DNA Sequences Using Evolutionary Information Based on Deep Learning. Cells 2019, 8, 1635. [Google Scholar] [CrossRef] [Green Version]
- Nazari, I.; Tayara, H.; Chong, K.T. Branch Point Selection in RNA Splicing Using Deep Learning. IEEE Access 2018, 7, 1800–1807. [Google Scholar] [CrossRef]
- Tahir, M.; Tayara, H.; Chong, K.T. irna-pseknc (2methyl): Identify rna 2′-o-methylation sites by convolution neural network and chou’s pseudo components. J. Theor. Biol. 2019, 465, 1–6. [Google Scholar] [CrossRef]
- Ali, S.D.; Alam, W.; Tayara, H.; Chong, K. Identification of Functional piRNAs Using a Convolutional Neural Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 1. [Google Scholar] [CrossRef]
- Tayara, H.; Tahir, M.; Chong, K.T. iss-cnn: Identifying splicing sites using convolution neural network. Chemom. Intell. Lab. Syst. 2019, 188, 63–69. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M. Machine learning based identification of protein—Protein interactions using derived features of physiochemical properties and evolutionary profiles. Artif. Intell. Med. 2017, 78, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Luan, S.; Nagai, L.A.E.; Su, R.; Zou, Q. Exploring sequencebased features for the improved prediction of DNA n4-methylcytosine sites in multiple species. Bioinformatics 2018, 35, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.D.; Kim, J.H.; Tayara, H.; Chong, K.T. Prediction of RNA 5-Hydroxymethylcytosine Modifications Using Deep Learning. IEEE Access 2021, 9, 8491–8496. [Google Scholar] [CrossRef]
- Jia, C.; Yang, Q.; Zou, Q. NucPosPred: Predicting species-specific genomic nucleosome positioning via four different modes of general PseKNC. J. Theor. Biol. 2018, 450, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Khanal, J.; Tayara, H.; Chong, K.T. Identifying Enhancers and Their Strength by the Integration of Word Embedding and Convolution Neural Network. IEEE Access 2020, 8, 58369–58376. [Google Scholar] [CrossRef]
- Jia, C.; He, W. EnhancerPred: A predictor for discovering enhancers based on the combination and selection of multiple features. Sci. Rep. 2016, 6, 38741. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Bari, A.G.; Reaz, M.R.; Choi, H.-J.; Jeong, B.-S. DNA encoding for splice site prediction in large DNA sequence. In International Conference on Database Systems for Advanced Applications; Springer: Berlin/Heidelberg, Germany, 2013; pp. 46–58. [Google Scholar]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Ying, X. An Overview of Overfitting and its Solutions. J. Phys. Conf. Ser. 2019, 1168, 022022. [Google Scholar] [CrossRef]
- Matthews, B. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. BBA Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Cai, L.; Ren, X.; Fu, X.; Peng, L.; Gao, M.; Zeng, X. iEnhancer-XG: Interpretable sequence-based enhancers and their strength predictor. Bioinformatics 2020, 37, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Yapp, E.K.Y.; Ho, Q.-T.; Nagasundaram, N.; Ou, Y.-Y.; Yeh, H.-Y. iEnhancer-5Step: Identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal. Biochem. 2019, 571, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.N.; Ibrahim, M.A.; Malik, M.I.; Dengel, A.; Ahmed, S. Enhancer-dsnet: A supervisedly prepared enriched sequence representation for the identification of enhancers and their strength. In International Conference on Neural Information Processing; Springer: Berlin/Heidelberg, Germany, 2020; pp. 38–48. [Google Scholar]
- Tan, K.K.; Le, N.Q.K.; Yeh, H.-Y.; Chua, M.C.H. Ensemble of Deep Recurrent Neural Networks for Identifying Enhancers via Dinucleotide Physicochemical Properties. Cells 2019, 8, 767. [Google Scholar] [CrossRef] [Green Version]
- Chicco, D. Ten quick tips for machine learning in computational biology. BioData Min. 2017, 10, 35. [Google Scholar] [CrossRef]
- Tianshun, G.; Qian, J. EnhancerAtlas 2.0: An updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2020, 48, D58–D64. [Google Scholar]
- Bo, Z.; Zhang, W.; Zhang, T.; Liu, B.; Jianga, J. Genome-Wide Prediction and Validation of Intergenic Enhancers in Arabidopsis Using Open Chromatin Signatures. Plant Cell 2015, 27, 2415–2426. [Google Scholar]
- Ivica, L.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer | Parameters |
|---|---|
| Input | - |
| Gaussian Noise | Standard deviation = 0.2 |
| Conv1D | Kernels = 128 |
| Kernel size = 7 | |
| Group Normalization | Groups = 2 |
| Max pooling | Window size = 2 |
| Dropout | Dropout rate = 0.2 |
| Conv1D | Kernels = 256 |
| Kernel size = 6 | |
| Group Normalization | Groups = 2 |
| Max pooling | Window size = 2 |
| Dropout | Dropout rate = 0.15 |
| Conv1D | Kernels = 256 |
| Kernel size = 5 | |
| Group Normalization | Groups = 2 |
| Max pooling | Window size = 2 |
| Dropout | Dropout rate = 0.04 |
| Flatten | - |
| Dense | Nodes = 16 |
| Output | Nodes = 1 |
| Chemical Properties | Class | Nucleotides |
|---|---|---|
| Ring Structure | Purine | [A, G] |
| Pyrimidine | [C, T] | |
| Hydrogen Bond | Strong | [C, G] |
| Weak | [A, T] | |
| Functional Group | Amino | [A, C] |
| Keto | [G, T] |
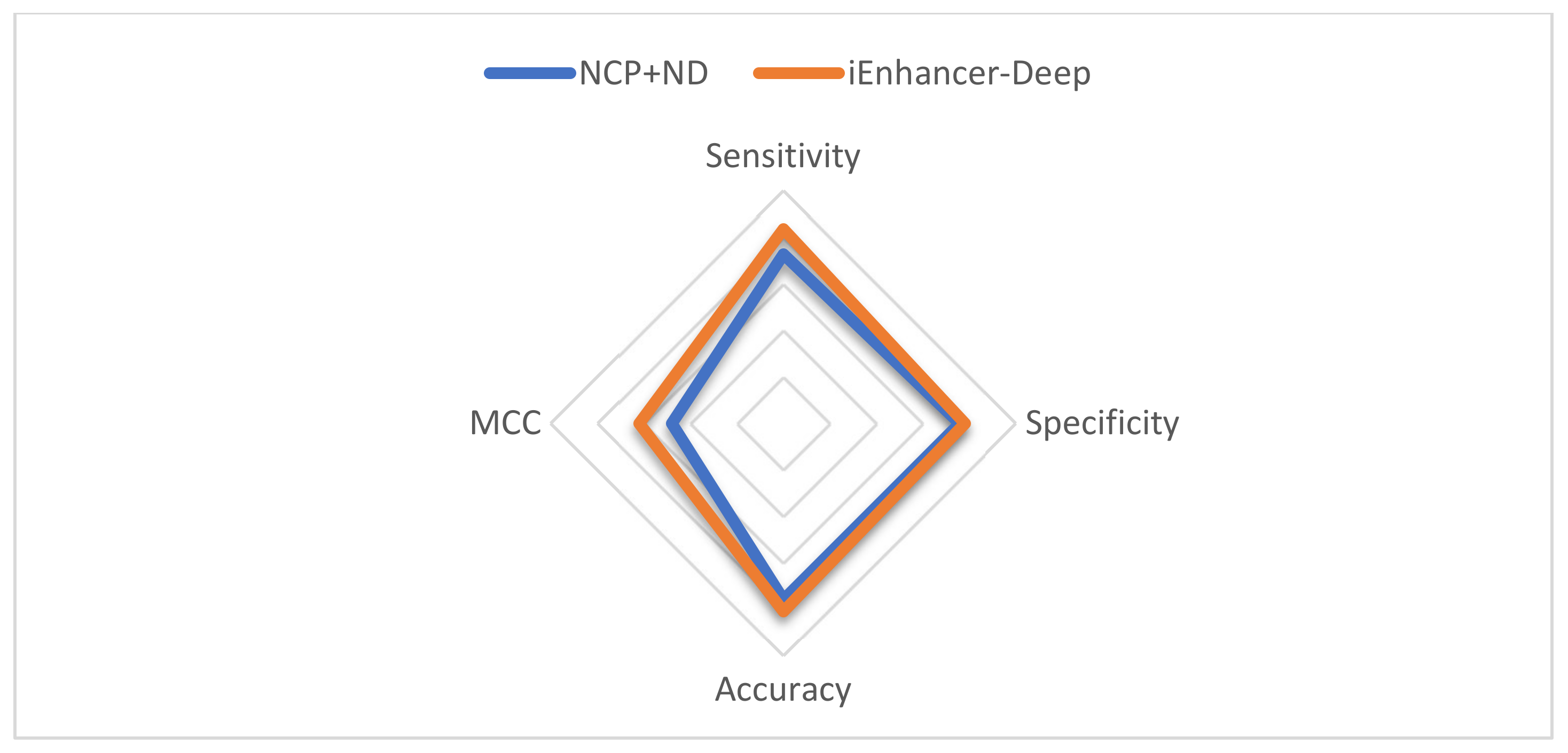
| Stage | Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|---|
| Stage 1 | NCP+ND | 72.7 | 72.7 | 74.59 | 0.49 |
| iEnhancer-Deep | 86.99 | 88.54 | 87.77 | 0.75 | |
| Stage 2 | NCP+ND | 72.6 | 75.9 | 75.9 | 0.48 |
| iEnhancer-Deep | 83.57 | 78.16 | 80.86 | 0.62 |
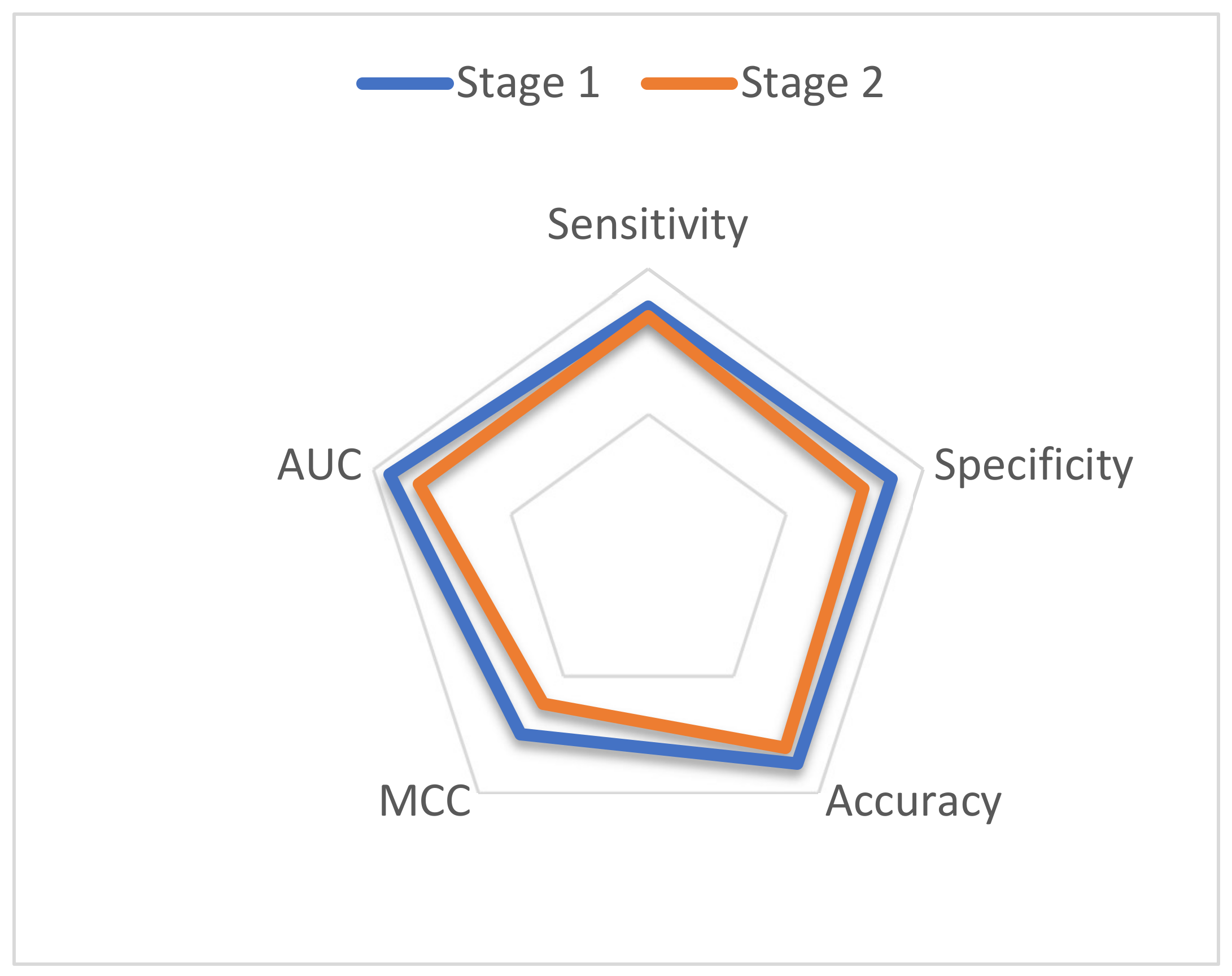
| Stages | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| Stage 1 | 86.99 | 88.54 | 87.77 | 0.75 |
| Stage 2 | 83.57 | 78.16 | 80.86 | 0.62 |
| Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| iEnhancer-EL | 75.67 | 80.39 | 78.03 | 0.56 |
| iEnhnacer-2L | 78.09 | 75.88 | 76.89 | 0.54 |
| EnhancerPred | 72.57 | 73.79 | 73.18 | 0.43 |
| iEnhancer-XG | 75.70 | 86.50 | 58.55 | 0.62 |
| iEnhancer-5Step | 81.10 | 83.50 | 82.30 | 0.65 |
| iEnhancer-DSNet | 76.00 | 76.00 | 76.00 | 0.52 |
| tan2019emsemble | 73.25 | 76.42 | 74.83 | .49 |
| iEnhancer-CNN | 75.88 | 88.88 | 80.63 | 0.69 |
| iEnhancer-Deep | 86.99 | 88.5 | 87.77 | 0.75 |
| Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| iEnhancer-EL | 69.00 | 61.05 | 65.03 | 0.31 |
| iEnhnacer-2L | 62.21 | 61.82 | 61.93 | 0.24 |
| EnhancerPred | 62. 67 | 61.46 | 62.06 | 0.24 |
| iEnhancer-XG | 74.94 | 58.55 | 66.74 | 0.33 |
| iEnhancer-5Step | 75.53 | 60.80 | 68.10 | 0.37 |
| iEnhancer-DSNet | 63.00 | 67.00 | 63.00 | 0.26 |
| tan2019emsemble | 79.65 | 38.28 | 58.96 | 0.19 |
| iEnhancer-CNN | 73.64 | 76.8 | 76.43 | 0.45 |
| iEnhancer-Deep | 83.57 | 78.16 | 80.86 | 0.62 |
| Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| iEnhancer-EL | 71.00 | 78.50 | 74.75 | 0.4969 |
| iEnhnacer-2L | 71.00 | 75.00 | 73.00 | 0.4604 |
| EnhancerPred | 73.50 | 74.50 | 74.00 | 0.4800 |
| iEnhancer-CNN | 78.25 | 79.00 | 77.50 | 0.5850 |
| iEnhancer-Deep | 81.5 | 67.00 | 74.02 | 0.4902 |
| Model | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| iEnhancer-EL | 54.00 | 68.00 | 61.00 | 0.2222 |
| iEnhnacer-2L | 47.00 | 74.00 | 60.50 | 0.2181 |
| EnhancerPred | 45.00 | 65.00 | 55.00 | 0.1021 |
| iEnhancer-CNN | 65.25 | 76.10 | 75.00 | 0.3232 |
| iEnhancer-Deep | 73.00 | 49.00 | 61.00 | 0.2266 |
| Model | Species | Total Enhancers | TP | FN |
|---|---|---|---|---|
| iEnhancer-CNN | M. musculus | 34,696 | 19,500 | 15,196 |
| iEnhancer-Deep | 22,184 | 12,512 | ||
| iEnhancer-CNN | D. melanogaster | 8060 | 4748 | 3312 |
| iEnhancer-Deep | 4511 | 3549 | ||
| iEnhancer-CNN | C. elegance | 11,870 | 4076 | 7794 |
| iEnhancer-Deep | 4381 | 7489 | ||
| iEnhancer-CNN | A. thaliana | 5871 | 462 | 5409 |
| iEnhancer-Deep | 855 | 5016 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamran, H.; Tahir, M.; Tayara, H.; Chong, K.T. iEnhancer-Deep: A Computational Predictor for Enhancer Sites and Their Strength Using Deep Learning. Appl. Sci. 2022, 12, 2120. https://doi.org/10.3390/app12042120
Kamran H, Tahir M, Tayara H, Chong KT. iEnhancer-Deep: A Computational Predictor for Enhancer Sites and Their Strength Using Deep Learning. Applied Sciences. 2022; 12(4):2120. https://doi.org/10.3390/app12042120
Chicago/Turabian StyleKamran, Haider, Muhammad Tahir, Hilal Tayara, and Kil To Chong. 2022. "iEnhancer-Deep: A Computational Predictor for Enhancer Sites and Their Strength Using Deep Learning" Applied Sciences 12, no. 4: 2120. https://doi.org/10.3390/app12042120
APA StyleKamran, H., Tahir, M., Tayara, H., & Chong, K. T. (2022). iEnhancer-Deep: A Computational Predictor for Enhancer Sites and Their Strength Using Deep Learning. Applied Sciences, 12(4), 2120. https://doi.org/10.3390/app12042120