
Exploring the Effect of the Number of Hydrogen Atoms on the Properties of Lanthanide Hydrides by DMFT



Abstract
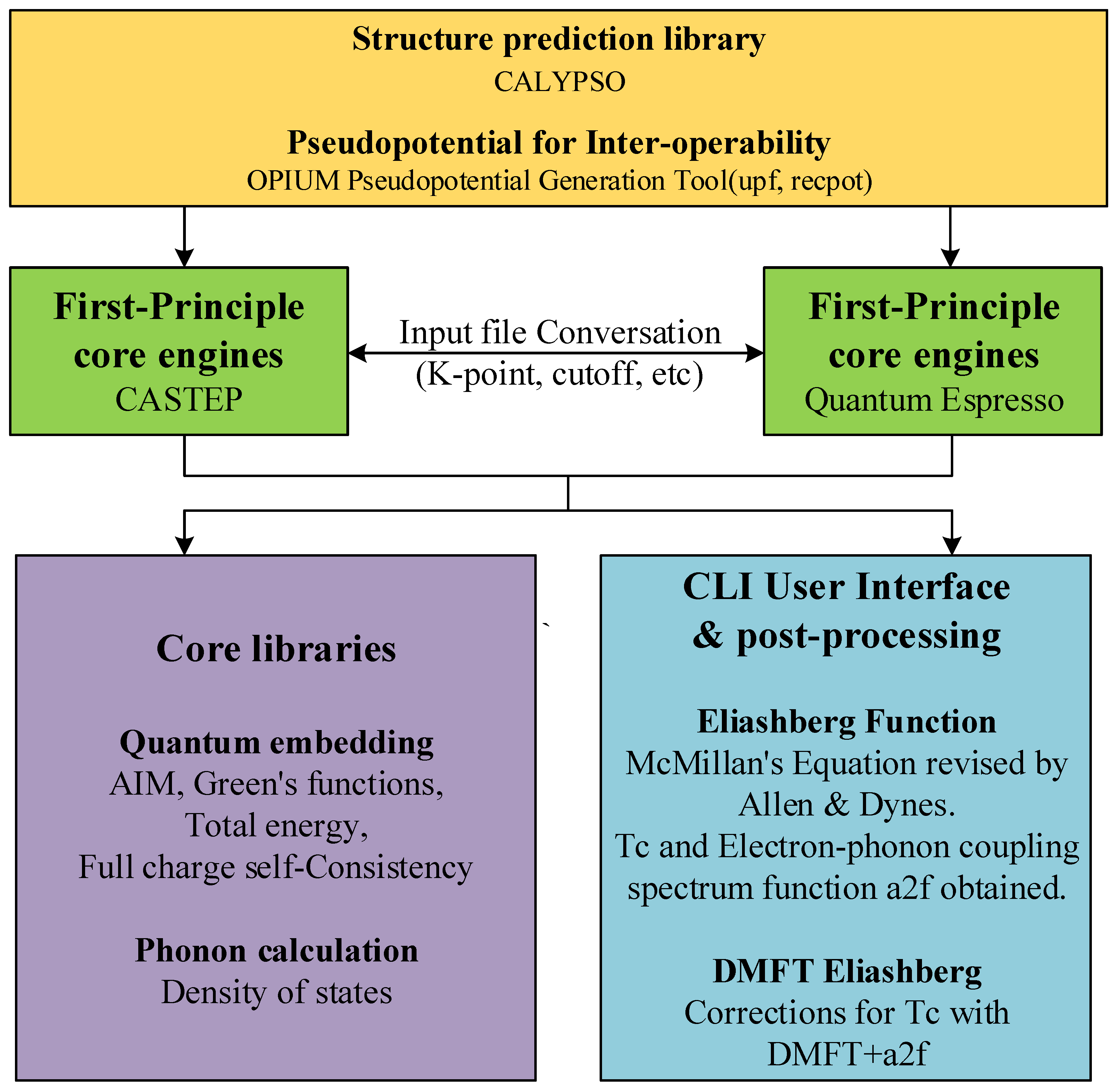
:1. Introduction
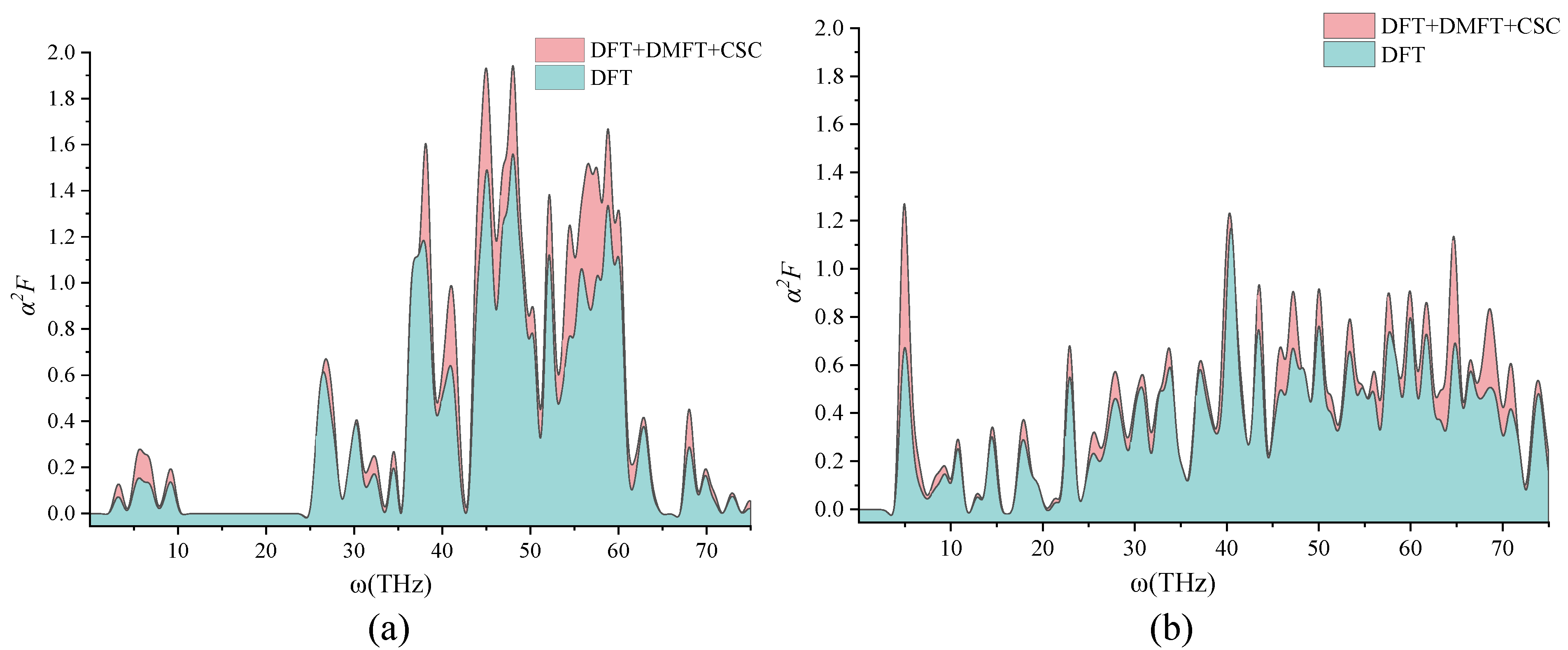
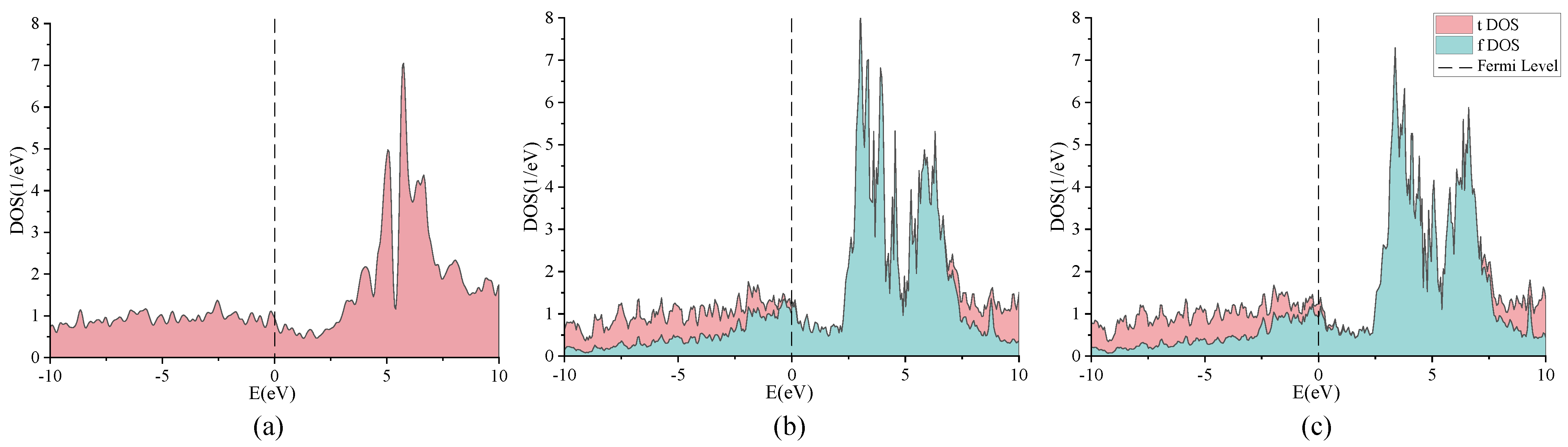
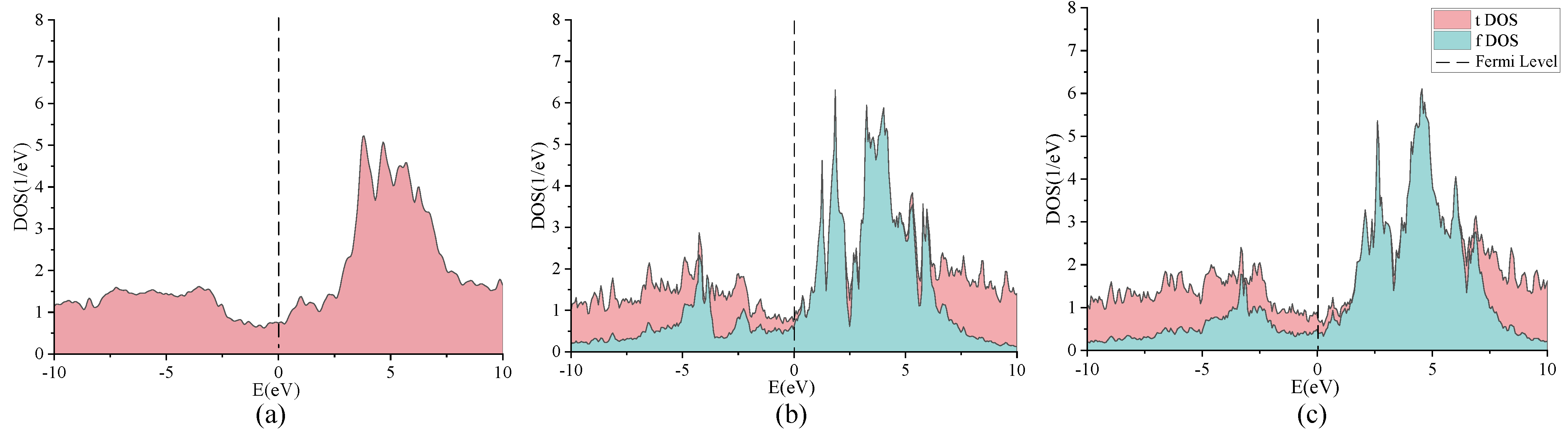
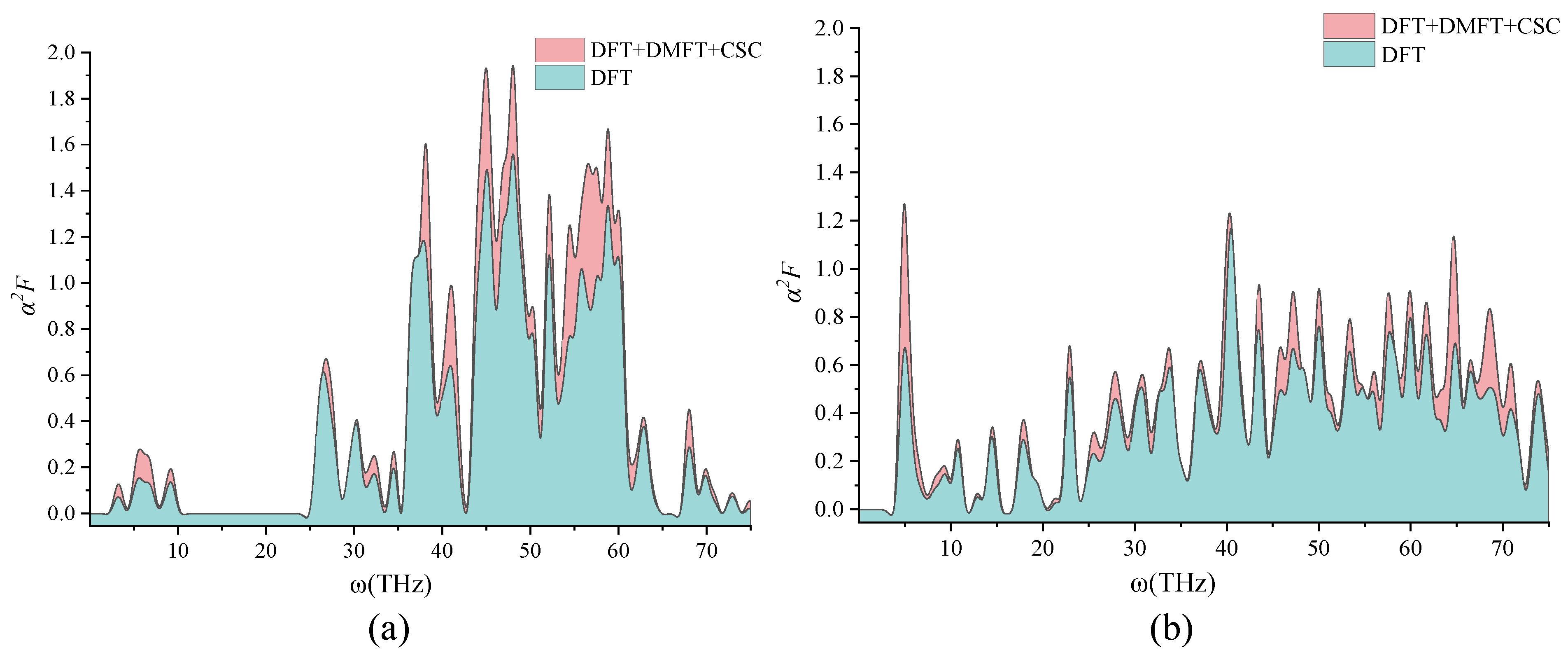
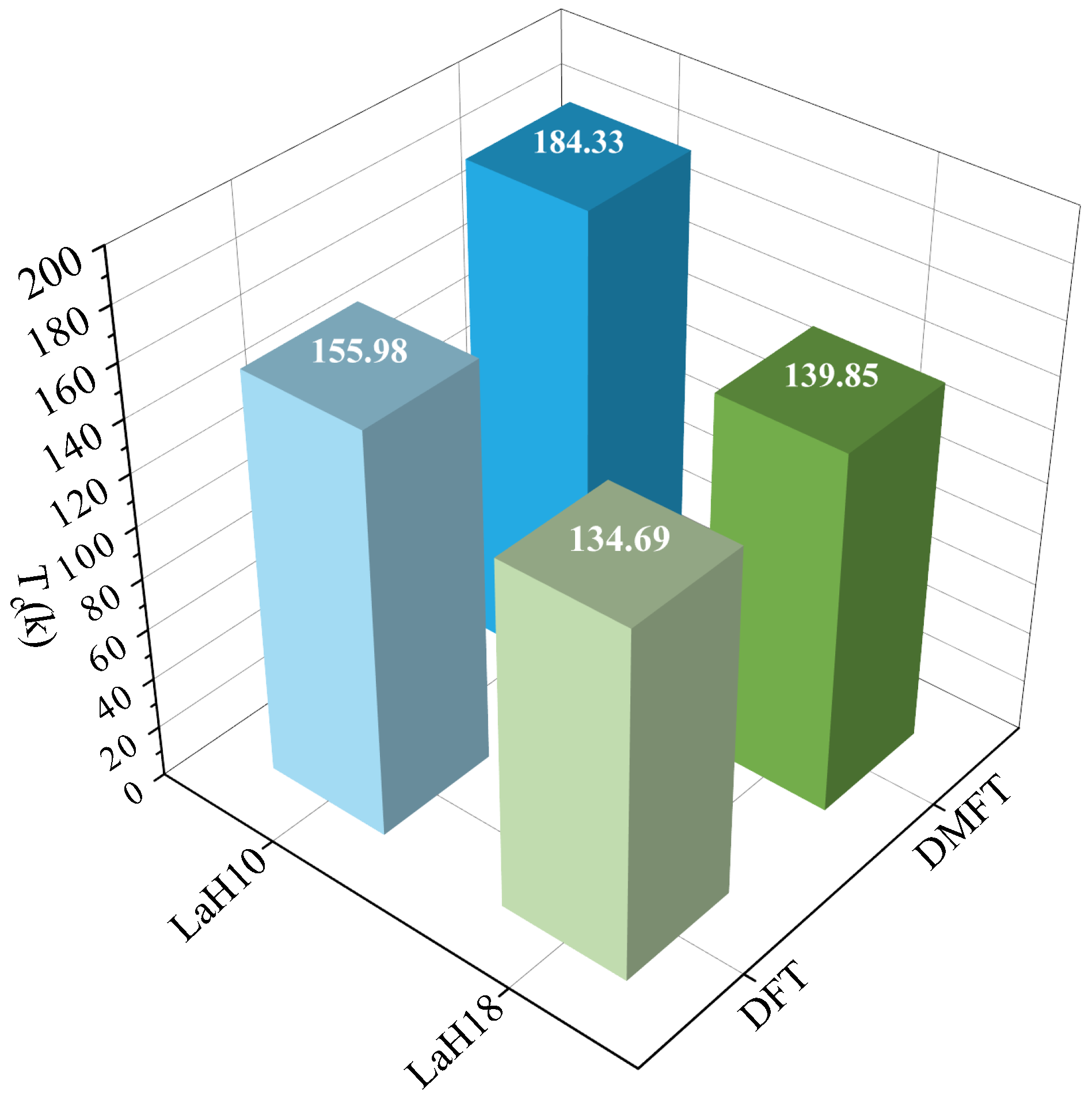
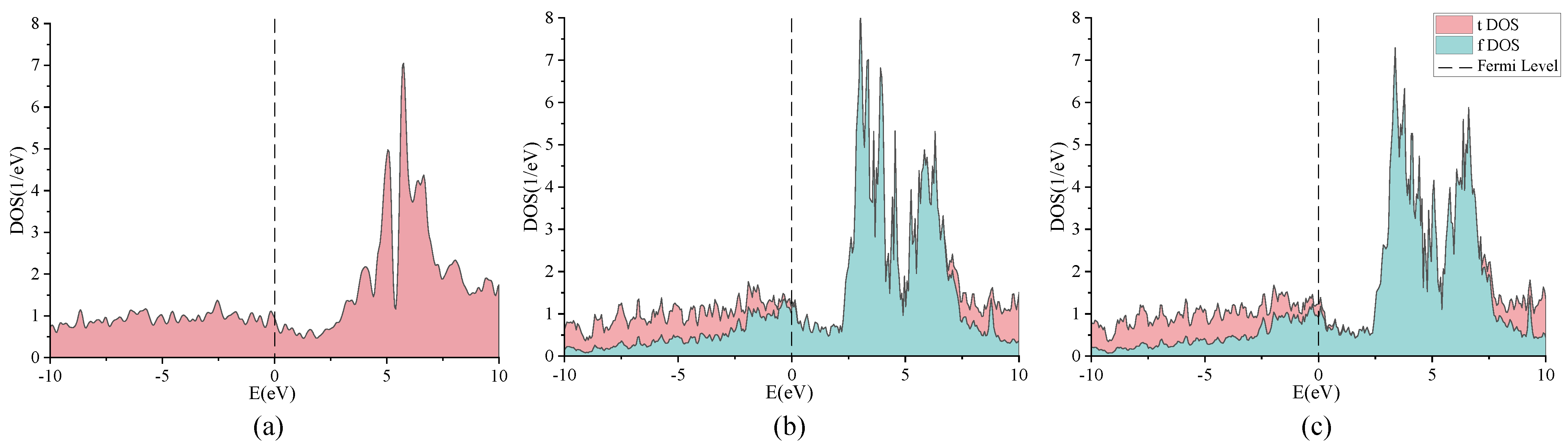
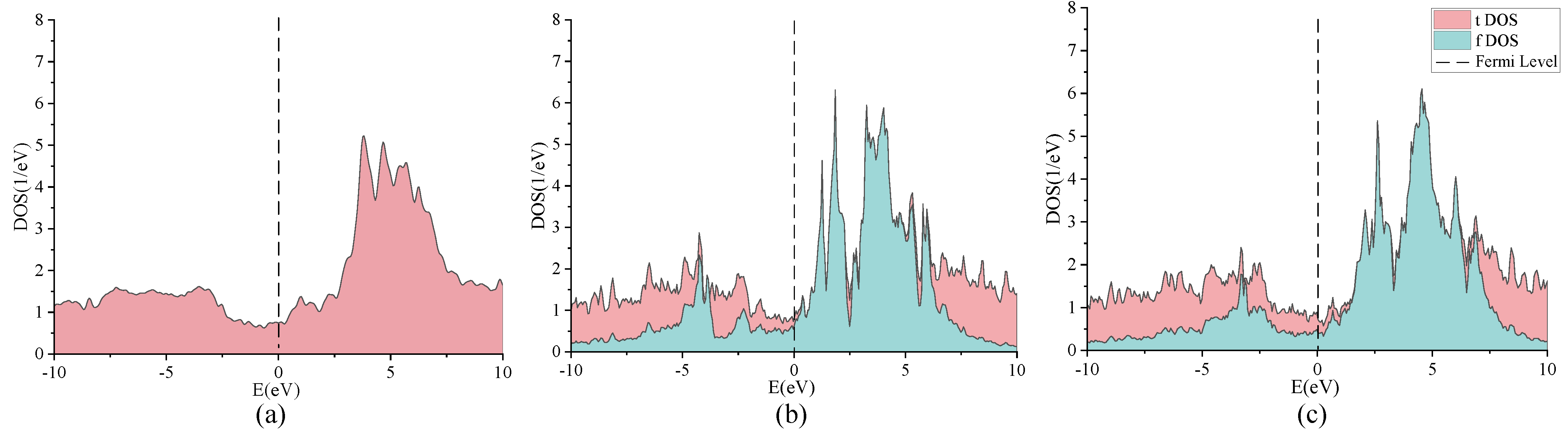
2. Discussion
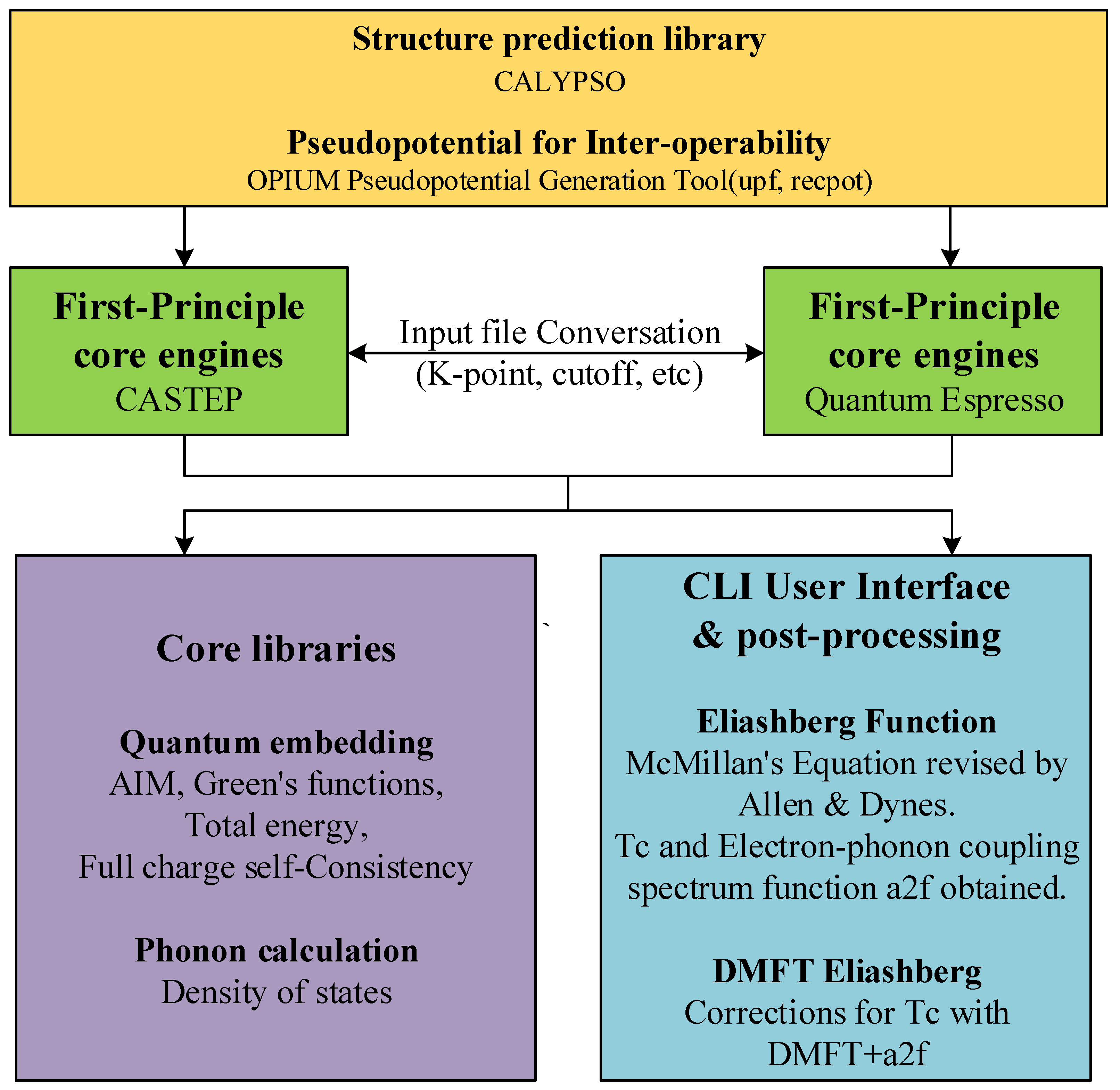
3. Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Space Group | Lattice Parameters (Å) | Atoms | Atomic Coordinates (Fractional) | |||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| LaH (400 GPa) | Fm-3m | a = b = c = 4.59461 = = = 90° | H(32f) | 0.62021 | 0.62021 | 0.62021 |
| H(8c) | 0.25000 | 0.25000 | 0.25000 | |||
| La(4a) | 0.00000 | 0.00000 | 0.00000 | |||
|
LaH (400 GPa) | Fmmm | a =5.78715 b = 7.06134 c = 3.33380 = = = 90° | H(16o) | 0.32905 | 0.27446 | 0.00000 |
| H(16l) | 0.08379 | 0.25000 | 0.25000 | |||
| H(16k) | 0.25000 | 0.38271 | 0.25000 | |||
| H(16o) | 0.33762 | 0.07756 | 0.00000 | |||
| La(8h) | 0.00000 | 0.61864 | 0.00000 | |||
| La(4a) | 0.00000 | 0.00000 | 0.00000 | |||
| Method | (States/eV/f.u.) | |||
|---|---|---|---|---|
| LaH | DFT | 1.35 | 1536 | 0.80 |
| DMFT | 1.76 | 1405 | 1.23 | |
| LaH | DFT | 1.65 | 1083 | 0.75 |
| DMFT | 2.22 | 909 | 0.83 |
References
- McMahon, J.M.; Ceperley, D.M. High-temperature superconductivity in atomic metallic hydrogen. Phys. Rev. B 2011, 84, 144515. [Google Scholar] [CrossRef] [Green Version]
- Borinaga, M.; Errea, I.; Calandra, M.; Mauri, F.; Bergara, A. Anharmonic effects in atomic hydrogen: Superconductivity and lattice dynamical stability. Phys. Rev. B 2016, 93, 174308. [Google Scholar] [CrossRef] [Green Version]
- Wigner, E.; Huntington, H. On the possibility of a metallic modification of hydrogen. J. Chem. Phys. 1935, 3, 764. [Google Scholar] [CrossRef]
- Cudazzo, P.; Profeta, G.; Sanna, A.; Floris, A.; Continenza, A.; Massidda, S.; Gross, E. Ab initio description of high-temperature superconductivity in dense molecular hydrogen. Phys. Rev. Lett. 2008, 100, 257001. [Google Scholar] [CrossRef] [Green Version]
- McMahon, J.M.; Morales, M.A.; Pierleoni, C.; Ceperley, D.M. The properties of hydrogen and helium under extreme conditions. Rev. Mod. Phys. 2012, 84, 1607. [Google Scholar] [CrossRef]
- Eremets, M.I.; Drozdov, A.P.; Kong, P.; Wang, H. Semimetallic molecular hydrogen at pressure above 350 gpa. Nat. Phys. 2019, 15, 1246. [Google Scholar] [CrossRef]
- Loubeyre, P.; Occelli, F.; Dumas, P. Synchrotron infrared spectroscopic evidence of the probable transition to metal hydrogen. Nature 2020, 577, 631. [Google Scholar] [CrossRef]
- Sun, W.; Kuang, X.; Keen, H.D.; Lu, C.; Hermann, A. Second group of high-pressure high-temperature lanthanide polyhydride superconductors. Phys. Rev. B 2020, 102, 144524. [Google Scholar] [CrossRef]
- Plekhanov, E.; Zhao, Z.; Macheda, F.; Wei, Y.; Bonini, N.; Weber, C. Computational materials discovery for lanthanide hydrides at high pressure for high temperature superconductivity. Phys. Rev. Res. 2022, 4, 013248. [Google Scholar] [CrossRef]
- Wang, C.; Liu, S.; Jeon, H.; Yi, S.; Bang, Y.; Cho, J.-H. Effect of hole doping on superconductivity in compressed ceh 9 at high pressures. Phys. Rev. B 2021, 104, L020504. [Google Scholar] [CrossRef]
- Li, B.; Miao, Z.; Ti, L.; Liu, S.; Chen, J.; Shi, Z.; Gregoryanz, E. Predicted high-temperature superconductivity in cerium hydrides at high pressures. J. Appl. Phys. 2019, 126, 235901. [Google Scholar] [CrossRef]
- Wei, Y.; Macheda, F.; Zhao, Z.; Tse, T.; Plekhanov, E.; Bonini, N.; Weber, C. High-temperature superconductivity in the lanthanide hydrides at extreme pressures. Appl. Sci. 2022, 12, 874. [Google Scholar] [CrossRef]
- Kruglov, I.A.; Semenok, D.V.; Song, H.; Szczsniak, R.; Wrona, I.A.; Akashi, R.; Esfahani, M.M.D.; Duan, D.; Cui, T.; Kvashnin, A.G.; et al. Superconductivity of lah 10 and lah 16 polyhydrides. Phys. Rev. B 2020, 101, 024508. [Google Scholar] [CrossRef] [Green Version]
- Kong, P.; Minkov, V.S.; Kuzovnikov, M.A.; Drozdov, A.P.; Besedin, S.P.; Mozaffari, S.; Balicas, L.; Balakirev, F.F.; Prakapenka, V.B.; Chariton, S.; et al. Superconductivity up to 243 k in the yttrium-hydrogen system under high pressure. Nat. Commun. 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Struzhkin, V.; Li, B.; Ji, C.; Chen, X.-J.; Prakapenka, V.; Greenberg, E.; Troyan, I.; Gavriliuk, A.; Mao, H.-K. Superconductivity in la and y hydrides: Remaining questions to experiment and theory. Matter Radiat. Extrem. 2020, 5, 028201. [Google Scholar] [CrossRef] [Green Version]
- Semenok, D.V.; Kvashnin, A.G.; Kruglov, I.A.; Oganov, A.R. Actinium hydrides ach10, ach12, and ach16 as high-temperature conventional superconductors. J. Phys. Chem. Lett. 2018, 9, 1920. [Google Scholar] [CrossRef] [Green Version]
- Semenok, D.V.; Kvashnin, A.G.; Ivanova, A.G.; Svitlyk, V.; Fominski, V.Y.; Sadakov, A.V.; Sobolevskiy, O.A.; Pudalov, V.M.; Troyan, I.A.; Oganov, A.R. Superconductivity at 161 k in thorium hydride thh10: Synthesis and properties. Mater. Today 2020, 33, 36. [Google Scholar] [CrossRef] [Green Version]
- Kvashnin, A.G.; Semenok, D.V.; Kruglov, I.A.; Wrona, I.A.; Oganov, A.R. High-temperature superconductivity in a th–h system under pressure conditions. ACS Appl. Mater. Interfaces 2018, 10, 43809. [Google Scholar] [CrossRef]
- Wang, H.; John, S.T.; Tanaka, K.; Iitaka, T.; Ma, Y. Superconductive sodalite-like clathrate calcium hydride at high pressures. Proc. Natl. Acad. Sci. USA 2012, 109, 6463. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Duan, D.; Ma, Y.; Yu, H.; Song, H.; Xie, H.; Li, D.; Tian, F.; Liu, B.; Cui, T. Unique phase diagram and superconductivity of calcium hydrides at high pressures. Inorg. Chem. 2019, 58, 2558. [Google Scholar] [CrossRef]
- Ma, L.; Wang, K.; Xie, Y.; Yang, X.; Wang, Y.; Zhou, M.; Liu, H.; Yu, X.; Zhao, Y.; Wang, H.; et al. High-Tc superconductivity in clathrate calcium hydride CaH6. arXiv 2021, arXiv:2103.16282. [Google Scholar]
- Li, Z.; He, X.; Zhang, C.; Zhang, S.; Feng, S.; Wang, X.; Yu, R.; Jin, C. Superconductivity above 200 K observed in superhydrides of calcium. arXiv 2021, arXiv:2103.16917. [Google Scholar]
- Szczesniak, R.; Durajski, A. Superconductivity well above room temperature in compressed mgh 6. Front. Phys. 2016, 11, 1. [Google Scholar] [CrossRef]
- Lonie, D.C.; Hooper, J.; Altintas, B.; Zurek, E. Metallization of magnesium polyhydrides under pressure. Phys. Rev. B 2013, 87, 054107. [Google Scholar] [CrossRef] [Green Version]
- Snider, E.; Dasenbrock-Gammon, N.; McBride, R.; Debessai, M.; Vindana, H.; Vencatasamy, K.; Lawler, K.V.; Salamat, A.; Dias, R.P. Room-temperature superconductivity in a carbonaceous sulfur hydride. Nature 2020, 586, 373. [Google Scholar] [CrossRef]
- Fu, Y.; Du, X.; Zhang, L.; Peng, F.; Zhang, M.; Pickard, C.J.; Needs, R.J.; Singh, D.J.; Zheng, W.; Ma, Y. High-pressure phase stability and superconductivity of pnictogen hydrides and chemical trends for compressed hydrides. Chem. Mater. 2016, 28, 1746. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Conway, L.; Sun, W.; Kuang, X.; Lu, C.; Hermann, A. Phase stability and superconductivity of lead hydrides at high pressure. Phys. Rev. B 2021, 103, 035131. [Google Scholar] [CrossRef]
- Fedorov, A.; Laubschat, C.; Starke, K.; Weschke, E.; Barholz, K.-U.; Kaindl, G. Surface shift of the unoccupied 4f state in la metal. Phys. Rev. Lett. 1993, 70, 1719. [Google Scholar] [CrossRef]
- Pfleiderer, C. Superconducting phases of f-electron compounds. Rev. Mod. Phys. 2009, 81, 1551. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Calypso: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.; Eberhart, R. Particle swarm optimization. In Proceedings of the 1995 IEEE International Conference on Neural Networks Proceedings, Perth, Australia, 27 November–1 December 1995; Volume 4. [Google Scholar]
- Eberhat, R.; Kennedy, J. A new optimizer using particle swarm theory. In Proceedings of the Sixth International Symposium on Micro Machine and Human Science, Piscataway, NJ, USA, 4–6 October 1995; pp. 39–43. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using castep. Z. Für Krist.-Cryst. Mater. 2005, 220, 567. [Google Scholar] [CrossRef] [Green Version]
- Plekhanov, E.; Hasnip, P.; Sacksteder, V.; Probert, M.; Clark, S.J.; Refson, K.; Weber, C. Many-body renormalization of forces in f-electron materials. Phys. Rev. B 2018, 98, 075129. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Plekhanov, E.; Blackbourn, D.; Acharya, S.; Weber, C. The mott to kondo transition in diluted kondo superlattices. Commun. Phys. 2019, 2, 1. [Google Scholar] [CrossRef]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227, Erratum in Phys. Rev. B 1991, 44, 13175. [Google Scholar] [CrossRef]
- Dynes, R. Mcmillan’s equation and the tc of superconductors. Solid State Commun. 1972, 10, 615. [Google Scholar] [CrossRef]
- Allen, P.B.; Dynes, R. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 1975, 12, 905. [Google Scholar] [CrossRef]
- Parcollet, O.; Ferrero, M.; Ayral, T.; Hafermann, H.; Krivenko, I.; Messio, L.; Seth, P. Triqs: A toolbox for research on interacting quantum systems. Comput. Phys. Commun. 2015, 196, 398. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Chachkarova, E.; Plekhanov, E.; Bonini, N.; Weber, C. Exploring the Effect of the Number of Hydrogen Atoms on the Properties of Lanthanide Hydrides by DMFT. Appl. Sci. 2022, 12, 3498. https://doi.org/10.3390/app12073498
Wei Y, Chachkarova E, Plekhanov E, Bonini N, Weber C. Exploring the Effect of the Number of Hydrogen Atoms on the Properties of Lanthanide Hydrides by DMFT. Applied Sciences. 2022; 12(7):3498. https://doi.org/10.3390/app12073498
Chicago/Turabian StyleWei, Yao, Elena Chachkarova, Evgeny Plekhanov, Nicola Bonini, and Cedric Weber. 2022. "Exploring the Effect of the Number of Hydrogen Atoms on the Properties of Lanthanide Hydrides by DMFT" Applied Sciences 12, no. 7: 3498. https://doi.org/10.3390/app12073498
APA StyleWei, Y., Chachkarova, E., Plekhanov, E., Bonini, N., & Weber, C. (2022). Exploring the Effect of the Number of Hydrogen Atoms on the Properties of Lanthanide Hydrides by DMFT. Applied Sciences, 12(7), 3498. https://doi.org/10.3390/app12073498