
Thermodynamics Modeling for Actinide Monocarbides and Mononitrides from First Principles


Abstract
1. Introduction
2. Computational Methods
2.1. Electronic-Structure Methods
2.2. Lattice-Dynamics Method
2.3. CALPHAD Method
3. Results
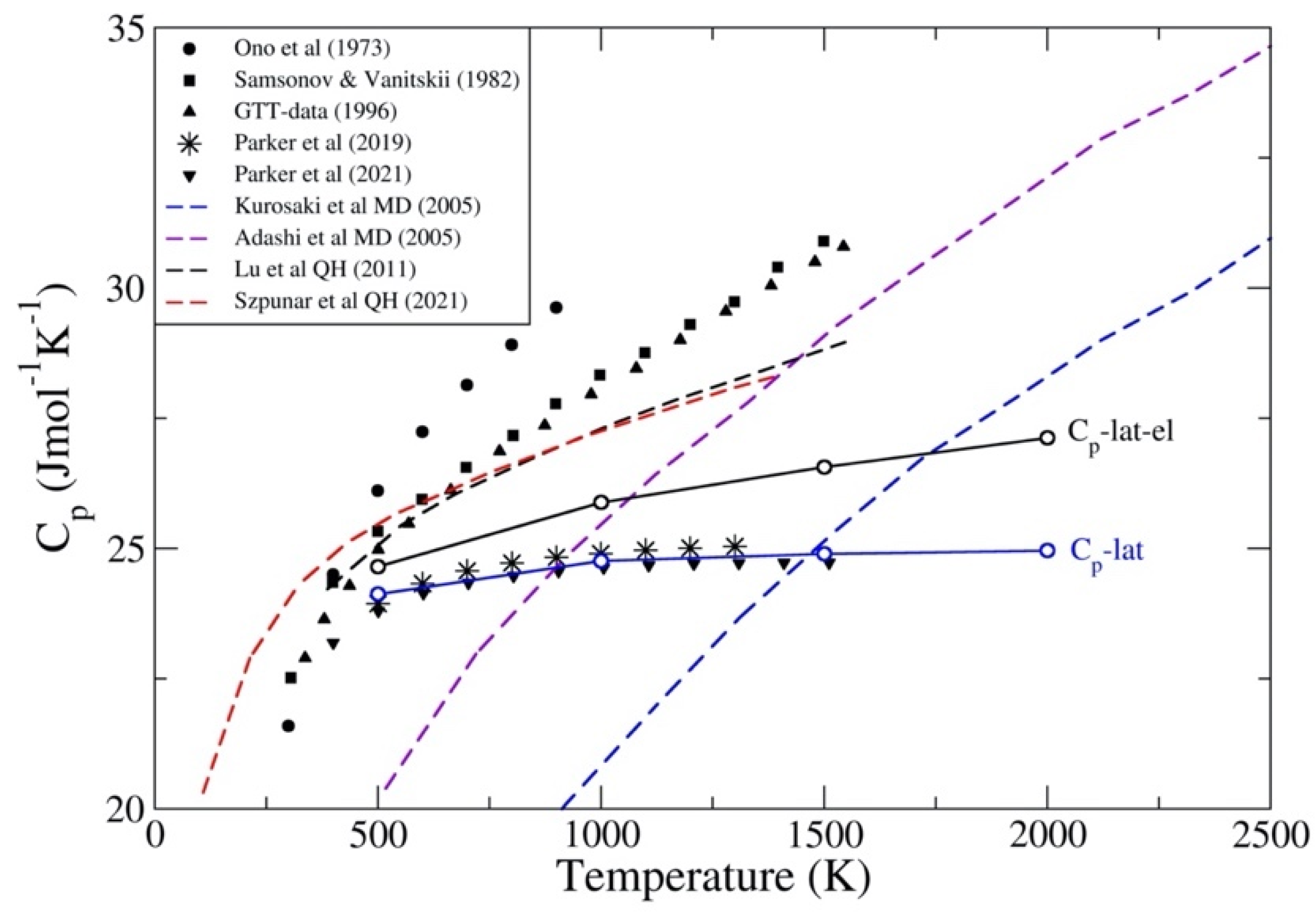
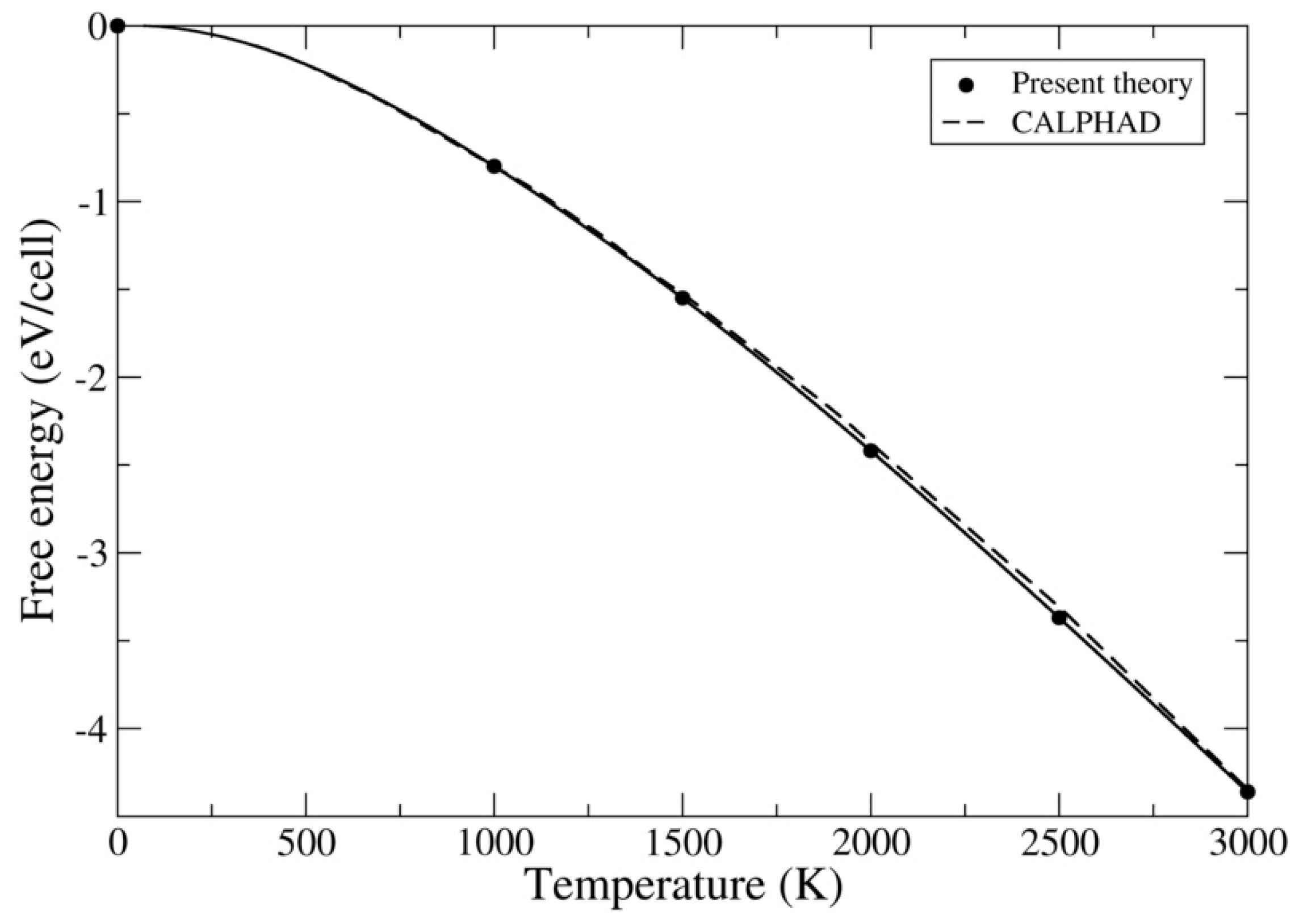
3.1. ThC and ThN
3.2. UC and UN
3.3. PuC and PuN
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abram, T.; Ion, S. Generation-IV nuclear power: A review of the state of the science. Eng. Policy 2008, 36, 4323–4330. [Google Scholar] [CrossRef]
- Matzke, H. Science of Advanced LMFBR Fuels; North-Holland: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Mishra, V.; Chaturvedi, S. Thermophysical properties of thorium compounds from first principles. In Proceedings of the Thorium energy conference 2015-ThEC15, Mumbai, India, 12–15 October 2015; Available online: http://www.thoriumenergyworld.com/uploads/6/9/8/7/69878937/thermophysical_properties_of_thorium_compounds_from_first_principles_thec15_paper.pdf (accessed on 1 December 2021).
- Sahafi, M.H.; Mahdavi, M. Ab-initio investigations on dynamical and lattice thermal behaviours of ThC. Bull. Mater. Sci. 2021, 44, 1–13. [Google Scholar] [CrossRef]
- Kývala, L.; Legut, D. Lattice dynamics and thermal properties of thorium metal and thorium monocarbide. Phys. Rev. B 2020, 101, 1–14. [Google Scholar] [CrossRef]
- Parker, S.S.; White, J.T.; Hosemann, P.; Nelson, A.T. Thermophysical properties of thorium mononitride from 298 to 1700 K. J. Nucl. Mater. 2019, 526, 1–9. [Google Scholar] [CrossRef]
- Parker, S.S.; Newman, S.; Fallgren, A.J.; White, J.T. Thermophysical properties of mixes of thorium and uranium nitride. JOM 2021, 73, 3564–3575. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Perron, A.; Sadigh, B.; Heo, T.W. Ground-state and thermodynamical properties of uranium mononitride from anharmonic first-principles theory. Appl. Sci. 2019, 9, 3914. [Google Scholar] [CrossRef]
- Kurosaki, K.; Adachi, J.; Uno, M.; Yamanaka, S. Molecular dynamics studies of actinide nitrides. J. Nucl. Mater. 2005, 344, 45–49. [Google Scholar] [CrossRef]
- Adashi, J.; Kurosaki, K.; Uno, M.; Yamanaka, S. A molecular dynamics study of thorium nitride. J. Alloys Compd. 2005, 394, 312–316. [Google Scholar] [CrossRef]
- Lu, Y.; Li, D.-F.; Wang, B.-T.; Li, R.-W.; Zhang, P. Electronic structures, mechanical and thermodynamical properties of ThN from first-principles calculations. J. Nucl. Mater 2011, 408, 136–141. [Google Scholar] [CrossRef]
- Szpunar, B.; Ranasinghe, J.I.; Malakkal, L.; Szpunar, J.A. First principles investigation of thermal properties of thorium mononitride. J. Alloys Compd. 2021, 879, 160467-1–160467-8. [Google Scholar] [CrossRef]
- Nakajima, K.; Arai, Y. Heat capacity of neptunium mononitride. J. Nucl. Sci. Technol. 2002, 39, 620–623. [Google Scholar] [CrossRef]
- Péres Daroca, D.; Jaroszewicz, S.; Llois, A.M.; Mosca, H.O. First-principles study of point defects in thorium carbide. J. Nucl. Mater. 2014, 454, 217–222. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Perron, A.; Moore, E.E.; Wu, C. Thermodynamics of plutonium monocarbide from anharmonic and relativistic theory. Appl. Sci. 2020, 10, 6524. [Google Scholar] [CrossRef]
- Lai, C.; Hu, Y.; Qiu, R. Thermodynamical stability of substoichiometric plutonium monocarbide from first-principles calculations. Phys. Chem. Chem. Phys. 2020, 22, 9009–9013. [Google Scholar] [CrossRef]
- Lai, C.; Hu, Y.; Qiu, R. Exploring the sub-stoichiometric behavior of plutonium mononitride. RSC Adv. 2020, 10, 24877–24881. [Google Scholar] [CrossRef]
- Kocevski, V.; Rehn, D.A.; Cooper, M.W.D.; Anderson, D.A. First-principles investigation of uranium mononitride (UN): Effect of magnetic ordering, spin-orbit interactions and exchange correlation functional. J. Nucl. Mater. 2021, 559, 153401. [Google Scholar] [CrossRef]
- Johansson, B.; Ahuja, R.; Eriksson, O.; Wills, J.M. Anomalous fcc crystal structure of thorium metal. Phys. Rev. Lett. 1995, 75, 280–283. [Google Scholar] [CrossRef]
- Söderlind, P. First-principles phase stability, bonding, and electronic structure of actinide metals. J. Electron Spectr. Rel. Phenom. 2014, 194, 2–7. [Google Scholar] [CrossRef]
- Wen, X.-D.; Martin, R.L.; Scuseria, G.E.; Rudin, S.P.; Batista, R.E. A screened hybrid DFT study of actinide oxides, nitrides, and carbides. J. Phys. Chem. C 2013, 117, 13122–13128. [Google Scholar] [CrossRef]
- Li, R.-S.; Tong, N.-H.; Wang, J.-T.; Xin, D.-Q.; Huang, S.-Q. A first principle calculation on electronic properties of plutonium mononitride: Insights from dynamical mean field theory. J. Nucl. Mater. 2018, 511, 277–283. [Google Scholar] [CrossRef]
- Wdowik, U.D.; Piekarz, P.; Legut, D.; Jaglo, G. Effect of spin-orbit and onsite Coulomb interactions on the electronic structure and lattice dynamics of uranium monocarbide. Phys. Rev. B 2016, 94, 054303-1–054303-8. [Google Scholar] [CrossRef]
- Eriksson, O.; Brooks, M.S.S.; Johansson, B. Orbital polarization in narrow-band systems: Application to volume collapses in light lanthanides. Phys. Rev. B 1990, 41, 7311–7314. [Google Scholar] [CrossRef] [PubMed]
- Eschrig, H.; Sargolzaei, M.; Koepernik, K.; Richter, M. Orbital polarization in Kohn-Sham-Dirac theory. EPL 2005, 72, 611–617. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Liechtenstein, A.I.; Terakura, K. Is Hund’s second rule responsible for the orbital magnetism in solids? Phys. Rev. Lett. 1998, 80, 5758–5761. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Sadigh, B. Density-functional theory for plutonium. Adv. Phys. 2019, 68, 1–47. [Google Scholar] [CrossRef]
- Sadigh, B.; Kutepov, A.; Landa, A.; Söderlind, P. Assessing relativistic effects and electron correlation in the actinide metals Th-Pu. Appl. Sci. 2019, 9, 5020. [Google Scholar] [CrossRef]
- Shein, I.R.; Ivanovskii, A.L. The influence of carbon non-stoichiometry on the electronic properties of thorium monocarbide ThC. Solid State Sci. 2010, 12, 1580–1584. [Google Scholar] [CrossRef]
- Wills, J.M.; Eriksson, O.; Andersson, P.; Delin, A.; Grechnyev, O.; Alouani, M. Full-Potential Electronic Structure Method; Springer Science and Business Media LLC: Berlin, Germany, 2010. [Google Scholar]
- Gerward, L.; Staun Olsen, J.; Benedict, U.; Itíe, J.-P.; Spirlet, J.C. Structural stability and equation of state of thorium carbide for pressures up to 36 GPa. J. Appl. Cryst. 1986, 19, 308–310. [Google Scholar] [CrossRef]
- Gerward, L.; Staun Olsen, J.; Benedict, U.; Itíe, J.-P.; Spirlet, J.C. The crystal structure and equation of state of thorium nitride for pressures up to 47 GPa. J. Appl. Cryst. 1985, 18, 339–341. [Google Scholar] [CrossRef]
- Vigier, N.; Auwer, D.; Fillaux, C.; Maslennikov, A.; Noël, H.; Roques, J.; Shuh, D.K.; Simoni, E.; Tyliszczak, T.; Moisy, P. New data on the structure of uranium monocarbide. Chem. Mater. 2008, 20, 3199–3204. [Google Scholar] [CrossRef]
- Staun Olsen, J.; Gerward, L.; Benedict, U. A new high-pressure phase of uranium nitride studied by X-ray diffraction and synchrotron radiation. J. Appl. Cryst. 1985, 18, 37–41. [Google Scholar] [CrossRef]
- Boeuf, A.; Caciuffo, R.; Fournier, J.M.; Manes, L.; Rebizant, J.; Roudaut, E.; Rustichelli, F. 239PuN powder neutron diffraction study. Solid State Comm. 1984, 52, 451–453. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, W.; Chen, L. Phase stability and mechanical properties of two new carbon crystals. EPL 2009, 87, 56003-1–56003-6. [Google Scholar] [CrossRef]
- Green, J.L.; Arnold, G.P.; Leary, J.A.; Nereson, N.G. Crystallographic and magnetic ordering studies of plutonium carbides using neutron diffraction. J. Nucl. Mater. 1970, 34, 281–289. [Google Scholar] [CrossRef]
- Curry, N.A. An investigation of the magnetic structure of uranium nitride by neutron diffraction. Proc. Phys. Soc. 1965, 86, 1193–1198. [Google Scholar] [CrossRef]
- Söderlind, P. Quantifying the importance of orbital over spin correlations in δ-Pu within density-functional theory. Phys. Rev. B 2008, 77, 1–5. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Souvatzis, P.; Eriksson, O.; Katsnelson, M.; Rudin, S. The self-consistent ab initio lattice dynamical method. Comput. Mater. Sci. 2009, 44, 888–894. [Google Scholar] [CrossRef]
- Kaufman, L.; Bernstein, H. Computer Calculation of Phase Diagrams with Special Reference to Refractory Metals; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Saunders, N.; Miodownik, A. CALPHAD Calculation of Phase Diagrams: A Comprehensive Guide; Elsevier Science: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Lukas, H.; Fries, S.; Sundman, B. Computational Thermodynamics: The CALPHAD Method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Ravat, B.; Oudot, B.; Perron, A.; Lalire, F.; Delaunay, F. Phase transformations in PuGa 1at.% alloy: Study of whole reversion process following martensitic transformation. J. Alloys Compd. 2013, 580, 298–309. [Google Scholar] [CrossRef]
- Perron, A.; Ravat, B.; Oudot, B.; Lalire, F.; Mouturat, K.; Delaunay, F. Phase transformations in Pu-Ga alloy: Synergy between simulations and experiments to elucidate direct and indirect reversion competition. Acta Mater. 2013, 61, 7109–7120. [Google Scholar] [CrossRef]
- Perron, A.; Turchi, P.; Landa, A.; Söderlind, P.; Ravat, B.; Oudot, B.; Delaunay, F.; Kurata, M. Thermodynamic re-assessment of the Pu-U system and its application to the ternary Pu-U-Ga system. J. Nucl. Mater. 2014, 454, 81–95. [Google Scholar] [CrossRef]
- Perron, A.; Turchi, P.; Landa, A.; Söderlind, P.; Ravat, B.; Oudot, B.; Delaunay, F. The Pu-U-Am system: An ab initio informed CALPHAD thermodynamic study. J. Nucl. Mater. 2015, 458, 425–441. [Google Scholar] [CrossRef]
- Moore, E.E.; Turchi, P.E.A.; Landa, A.; Söderlind, P.; Oudot, B.; Belof, J.L.; Stout, S.A.; Perron, A. Development of a CALPHAD thermodynamic database for Pu-U-Fe-Ga alloys. Appl. Sci. 2019, 9, 5040. [Google Scholar] [CrossRef]
- Grimvall, G. Spin disorder in paramagnetic fcc iron. Phys. Rev. B 1989, 39, 12300–12301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Classical mean-field approach for thermodynamics: Ab initio thermophysical properties of cerium. Phys. Rev. B 2000, 61, R11863–R11866. [Google Scholar] [CrossRef]
- Danan, J. Chaleur specifique de 2 a 300 K monocarbure de thorium. J. Nucl. Mater. 1975, 57, 280–282. [Google Scholar] [CrossRef]
- Grimvall, G.; Häglund, J.; Fernández Guillermet, A. Spin fluctuations in paramagnetic chromium determined from entropy considerations. Phys. Rev. B 1993, 47, 15338–15341. [Google Scholar] [CrossRef]
- Ono, F.; Kanno, M.; Mukaibo, T. Heat capacity of thorium nitrides from 450 to 800K. J. Nucl. Sci. Technol. 1973, 10, 391–395. [Google Scholar] [CrossRef][Green Version]
- Samsonov, G.V.; Vanitskii, I.M. Handbook of Refractory Metals; Plenum Publishing Corporation: New York, NY, USA, 1982. [Google Scholar]
- The SGTE Pure Substance and Dolution Database, GTT-Data Services. 1996. Available online: http://www.thermocalc.com/TCDATA.htm (accessed on 1 December 2021).
- Guéneau, C.; Dupin, S.; Sundman, B.; Martial, C.; Dumas, J.-C.; Gossé, S.; Chatain, S.; De Bruycker, F.; Manara, D.; Konings, R.J.M. Thermodynamic modelling of advanced oxide and carbide nuclear fuels: Description of the U-Pu-O-C systems. J. Nucl. Mater. 2011, 419, 145–167. [Google Scholar] [CrossRef]
- Chevalier, P.-Y.; Fischer, E.; Cheynet, B. Thermodynamic modelling of the N-U system. J. Nucl. Mater. 2000, 280, 136–150. [Google Scholar] [CrossRef]
- Besmann, T.; Shin, D.; Lindemer, T.B. Uranium nitride as LWR TRISO fuel: Thermodynamic modeling of U-C-N. J. Nucl. Mater. 2012, 427, 162–168. [Google Scholar] [CrossRef]
- OECD NEA/NSC: Thermodynamics of Advanced Fuels–International Database (TAF-ID), (n.d.). Available online: https://www.oecd-nea.org/science/taf-id/taf-id-public/ (accessed on 7 November 2019).
- DeCresente, M.A.; Miller, A.D. High temperature properties of uranium carbide. In Carbides in Nuclear Energy; Russel, L.R., Ed.; Macmillan: London, UK, 1964; Volume 1, pp. 342–357. [Google Scholar]
- Moser, J.B.; Kruger, O.L. Thermal conductivity and heat capacity of the monocarbide, monophosphide, and monosulfide of uranium. J. Appl. Phys. 1967, 38, 3215–3222. [Google Scholar] [CrossRef]
- Affortit, C. Chaleur specifique de UC et UN. J. Nucl. Mater. 1970, 34, 105–107. [Google Scholar] [CrossRef]
- Oetting, F.L.; Navratil, J.D.; Storms, E.K. The chemical thermodynamic properties of nuclear materials: (II) High temperature enthalpy of the uranium carbides. J. Nucl. Mater. 1973, 45, 271–283. [Google Scholar] [CrossRef]
- Westrum, E.G.; Barber, C.M. Uranium mononitride: Heat capacity and thermodynamic properties from 5 to 350 K. J. Chem. Phys. 1966, 45, 635–638. [Google Scholar] [CrossRef]
- Conway, J.B.; Flagella, P.N. Physical and Mechanical Properties of Reactor Materials; GEMP-1012; General Electric Company: Cincinnati, OH, USA, 1969. [Google Scholar]
- Fulkerson, W.; Kollie, T.G.; Weawer, S.C.; Moore, J.P.; Williams, R.K. Plutonium 1970 and other Actinides Part I and II. In Proceedings of the 4th International Conference on Plutonium and other Actinides (AIME), Santa Fe, NM, USA, 5–9 October 1970. [Google Scholar]
- Takahasi, Y.; Murabayashi, M.; Akimoto, Y.; Mukaibo, T. Uranium mononitride: Heat capacity and thermal conductivity from 298 to 1000 K. J. Nucl. Mater. 1971, 38, 303–308. [Google Scholar] [CrossRef]
- Oetting, F.L.; Leitnaker, J.M. The chemical thermodynamic properties of nuclear materials I. Uranium mononitride. J. Chem. Thermodyn. 1972, 4, 199–211. [Google Scholar] [CrossRef]
- Cordfunke, E.H.P.; Muis, R.P. The heat capacity of uranium mononitride. J. Nucl. Mater. 1972, 42, 233–234. [Google Scholar] [CrossRef]
- Matsui, T.; Ohse, R.W. Thermodynamic properties of uranium nitride, plutonium nitride and uranium-plutonium mixed nitride. High Temp. High Press. 1988, 20, 169–175. [Google Scholar]
- Söderlind, P.; Zhou, F.; Landa, A.; Klepeis, J.E. Phonon and magnetic structure in δ-plutonium from density-functional theory. Sci. Rep. 2015, 5, 15958-1–15958-6. [Google Scholar] [CrossRef] [PubMed]
- Kruger, O.L.; Savage, H. Heat capacity of plutonium monocarbide from 400° to 1300° K. J. Chem. Phys. 1964, 40, 3324–3328. [Google Scholar] [CrossRef]
- Oetting, F.L. The chemical thermodynamics of nuclear materials. J. Nucl. Mater. 1980, 88, 265–272. [Google Scholar] [CrossRef]
- Holley, C.E.; Rand, M.H.; Storms, E.K. The Chemical Thermodynamics of Actinide Elements and Compounds. Part 6: The Actinide Carbides; IAEA: Vienna, Austria, 1984; pp. 53–72. [Google Scholar]
- Spear, K.E.; Leitnaker, J.M. A consistent set of thermodynamic values for plutonium mononitride. J. Am. Ceram. Soc. 1968, 51, 706–709. [Google Scholar] [CrossRef]
- Oetting, F.L. The chemical thermodynamic properties of nuclear materials III: Plutonium mononitride. J. Chem. Thermodyn. 1978, 10, 941–948. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | ThC | ThN | UC | UN | PuC | PuN |
|---|---|---|---|---|---|---|
| Theory-NM | 19.20 | 17.27 | 15.24 | 14.53 | 14.71 | 14.70 |
| Theory-AF | – | – | 15.27 | 14.85 | 16.00 | 15.00 |
| Theory-FM | – | – | 15.33 | 14.85 | 16.19 | 15.18 |
| Experiment | 18.84 | 17.41 | 15.26 | 14.62 | 15.44 | 14.89 |
| Magnetic Moment | UC | UN | PuC | PuN |
|---|---|---|---|---|
| Spin | −0.04 | −1.31 | −3.81 | 4.10 |
| Orbital | 0.08 | 2.09 | 3.92 | −3.60 |
| Total | 0.04 | 0.78 | 0.11 | 0.50 |
| Property | ThC | ThN | UC | UN | PuC | PuN |
|---|---|---|---|---|---|---|
| Atomic volume | 19.25 | 17.52 | 15.55 | 14.91 | 15.87 | 15.10 |
| Spin moment | – | – | 0.685 | 1.246 | 3.571 | 3.869 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Söderlind, P.; Moore, E.E.; Wu, C.J. Thermodynamics Modeling for Actinide Monocarbides and Mononitrides from First Principles. Appl. Sci. 2022, 12, 728. https://doi.org/10.3390/app12020728
Söderlind P, Moore EE, Wu CJ. Thermodynamics Modeling for Actinide Monocarbides and Mononitrides from First Principles. Applied Sciences. 2022; 12(2):728. https://doi.org/10.3390/app12020728
Chicago/Turabian StyleSöderlind, Per, Emily E. Moore, and Christine J. Wu. 2022. "Thermodynamics Modeling for Actinide Monocarbides and Mononitrides from First Principles" Applied Sciences 12, no. 2: 728. https://doi.org/10.3390/app12020728
APA StyleSöderlind, P., Moore, E. E., & Wu, C. J. (2022). Thermodynamics Modeling for Actinide Monocarbides and Mononitrides from First Principles. Applied Sciences, 12(2), 728. https://doi.org/10.3390/app12020728