
Optimization of Protocol for Construction of Fungal ITS Amplicon Library for High-Throughput Illumina Sequencing to Study the Mycobiome of Aspen Leaves





Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Materials
2.2. DNA Extraction
2.3. Primers, PCRs, and Gel Electrophoresis
3. Results and Discussions
3.1. Choice of Extraction Kit for Fungal DNA Extraction
3.2. Optimization of Fungal Primer Pairs
3.3. Optimization of Extension PCR
3.4. Confirmation of Indexing for Illumina Library Sequencing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cordier, T.; Alonso-Sáez, L.; Gentil, L.; Aylagas, E.; Bohan, D.A.; Bouchez, A.; Chariton, A.; Creez, S.; Frühe, L.; Keck, F.; et al. Ecosystem motnitoring powered by environmental genomics: A review of current stretegies with an implementation roadmap. Mol. Ecol. 2019, 30, 2937–2958. [Google Scholar] [CrossRef]
- Williams, A.N.; Stavrinides, J. A microbial sampling and community reconstruction activity for introducing students to the burgeoning field of metagenomics. J. Microb. Biol. Educ. 2020, 21, 80. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2018, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Bálint, M.; Tiffin, P.; Hallström, B.; O’Hara, R.B.; Olson, M.S.; Fankhauser, J.D.; Piepenbring, M.; Schmit, I. Host Genotype Shapes the Foliar Fungal Microbiome of Balsam Poplar (Populus balsamifera). PLoS ONE 2013, 8, e53987. [Google Scholar] [CrossRef] [Green Version]
- Cordier, T.; Robin, C.; Capdevielle, X.; Loustau, M.; Vacher, C. Spatial variability of phyllosphere fungal assemblages: Genetic distance predominates over geographic distance in a European beech stand (Fagus sylvatica). Fungal Ecol. 2012, 5, 509–520. [Google Scholar] [CrossRef]
- Siddique, A.B.; Biella, P.; Unterseher, M.; Albrectsen, B.R. Mycobiome of young beech trees are distinguished by organ rather than habitat, and community analyses suggest competitive interactions among twig fungi. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Cordier, T.; Robin, C.; Capdevielle, X.; Fabreguettes, O.; Desprez-Loustau, M.L.; Vacher, C. The composition of phyllosphere fungal assemblages of european beech (Fagus sylvatica) varies significantly along an elevation gradient. New Phytol. 2012, 196, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Johansson, A.M.; Ahlinder, J.; Lundkvist, M.J.; Singh, N.J.; Brodin, T.; Forsman, M.; Stenberg, P. Airborne microbial biodiversity and seasonality in northern and southern sweden. PeerJ 2020, 8, e8424. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.B.; Unterseher, M. A cost-effective and efficient strategy for Illumina sequencing of fungal communities: A case study of beech endophytes identified elevation as main explanatory factor for diversity and community composition. Fungal Ecol. 2016, 20, 175–185. [Google Scholar] [CrossRef]
- Hashizume, Y.; Sahashi, N.; Fukuda, K. The influence of altitude on endophytic mycobiota in Quercus acuta leaves collected in two areas 1000 km apart. For. Pathol. 2008, 38, 218–226. [Google Scholar] [CrossRef]
- Eusemann, P.; Schnittler, M.; Nilsson, R.H.; Jumpponen, A.; Dahl, M.B.; Wurth, D.G.; Buras, A.; Wilmking, M.; Unterseher, M. Habitat conditions and phenological tree traits overrule the influence of tree genotype in the needle mycobiome-Picea glauca system at an arctic treeline ecotone. New Phytol. 2016, 211, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Würth, D.G.; Dahl, M.B.; Trouillier, M.; Wilmking, M.; Unterseher, M.; Scholler, M.; Sørensen, S.; Mortensen, M.; Schnittler, M. The needle mycobiome of Picea glauca—A dynamic system reflecting surrounding environment and tree phenological traits. Fungal Ecol. 2019, 41, 177–186. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Nilsson, R.H.; Tedersoo, L.; Abarenkov, K.; Carlsen, T.; Kjøller, R.; Kõljalg, U.; Pennanen, T.; Rosendahl, S.; Stenlid, J.; et al. Fungal community analysis by high-throughput sequencing of amplified markers—A user’s guide. New Phytol. 2013, 199, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldrian, P.; Vetrovsky, T.; Lepinay, C.; Kohout, P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers. 2021, 1–9. [Google Scholar] [CrossRef]
- Angebault, C.; Payen, M.; Woerther, P.L.; Rodriguez, C.; Botterel, F. Combined bacterial and fungal targeted amplicon sequencing of respiratory samples: Does the DNA extraction method matter? PLoS ONE 2020, 15, e0232215. [Google Scholar] [CrossRef]
- Clemmensen, K.E.; Ihrmark, K.; Durling, M.B.; Lindahl, B.D. Sample Preparation for Fungal Community Analysis by High-Throughput Sequencing of Barcode Amplicons. In Microbial Environmental Genomics (MEG); Martin, F., Uroz, S., Eds.; Methods in Molecular Biology Humana Press: New York, NY, USA, 2016; Volume 1399, pp. 61–88. [Google Scholar] [CrossRef]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate Estimation of Fungal Diversity and Abundance through Improved Lineage-Specific Primers Optimized for Illumina Amplicon Sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandau, F.; Albrectsen, B.R.; Robinson, K.M.; Gundale, M.J. European aspen with high compared to low constitutive tannin defenses grow taller in response to anthropogenic nitrogen enrichment. For. Ecol. Manag. 2021, 487, 118985. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification, and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. A Guide Methods Appl. 1990, 18, 315–322. [Google Scholar] [CrossRef]
- Fahle, G.A.; Fischer, S.H. Comparison of six commercial DNA extraction kits for recovery of cytomegalovirus DNA from spiked human specimens. J. Clin. Microbiol. 2000, 38, 3860–3863. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.J.; Wang, Y.; Wang, Q.; Han, Y.C.; Hu, Y.J. Comparison of three methods for isolation of nucleic acids from membranate inner ear tissue of rats. China Med. J. 2006, 119, 986–990. [Google Scholar] [CrossRef]
- Psifidi, A.; Dovas, C.I.; Bramis, G.; Lazou, T.; Russel, C.L.; Arsenos, G. Comparison of Eleven Methods for Genomic DNA Extraction Suitable for Large-Scale Whole-Genome Genotyping and Long-Term DNA Banking Using Blood Samples. PLoS ONE 2015, 10, e0178215. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Nikkila, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilic-Stojanovic, M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.G. Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol. 1997, 63, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Zhang, T. Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl. Microbiol. Biotechnol. 2013, 97, 4607–4616. [Google Scholar] [CrossRef] [Green Version]
- Free, S.J. Fungal Cell Wall Organization and Biosynthesis. Adv. Genet. 2013, 81, 33–82. [Google Scholar] [CrossRef]
- Psifidi, A.; Dovas, C.I.; Banos, G. A comparison of six methods for genomic DNA extraction suitable for PCR-based genotyping applications using ovine milk samples. Mol. Cell. Probes 2010, 24, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrie, R.M.; Schwarz, M.J.; Robertson, N.H.; Vaudin, S.; Super, M.; Malone, G.; Little, S. Development, multiplexing, and application of ARMS tests for common mutations in the CFTR gene. Am. J. Hum. Genet. 1992, 51, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Chumakov, K.M. Reverse-transcriptase can inhibit PCR and stimulate primer-dimer formation. PCR Methods Appl. 1994, 4, 62–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownie, J.; Shawcross, S.; Theaker, J. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 1997, 25, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Mohapatra, S.C.; Hsu, J.T. Studies on primer-dimer formation in polymerase chain reaction (PCR). Biotechnol. Tech. 1999, 13, 43–46. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Paepe, A.; Speleman, F. Elimination of primer-dimer artifacts and genomic coamplification using a two-step SYBR green I real time RT-PCR. Anal. Biochem. 2002, 303, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.T.; Kim, Y.J.; Kim, S.H. Annealing control primer system for improving specificity of PCR amplification. Biotechniques 2003, 35, 80–84. [Google Scholar] [CrossRef]
- Yang, Z.; Le, J.; Hutter, D.; Bradley, K.M.; Overton, B.R.; Lendon, C.; Benner, S. Eliminating primer dimers and improving SNP detection using self-avoiding molecular recognition system. Biol. Methods Protoc. 2020, 5, bpaa004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loquez, V.; Hall, D.; ALbrectsen, B.R.; Karlsson, J.; Ingvarsson, P.; Jansson, S. Natural phenological variation in aspen (Populus tremula): The SwAsp collection. Tree Genet. Genomes 2008, 4, 279–292. [Google Scholar] [CrossRef]
- Berry, D.; Ben Mahfoudh, K.; Wagner, M.; Loy, A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 2011, 77, 46–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Wen, C.; Qin, Y. Phasing amplicon sequencing on Illumina Miseq for robust environmental microbial community analysis. BMC Microbiol. 2015, 15, 125. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012, 40, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, R.H.; Hildebrand, F.; et al. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Guenay-Greunke, Y.; Bohan, D.A.; Traugott, M. Handling of targeted amplicon sequencing data focusing on index hopping and demultiplexing using a nested metabarcoding approach in ecology. Sci. Rep. 2021, 11, 19510. [Google Scholar] [CrossRef]
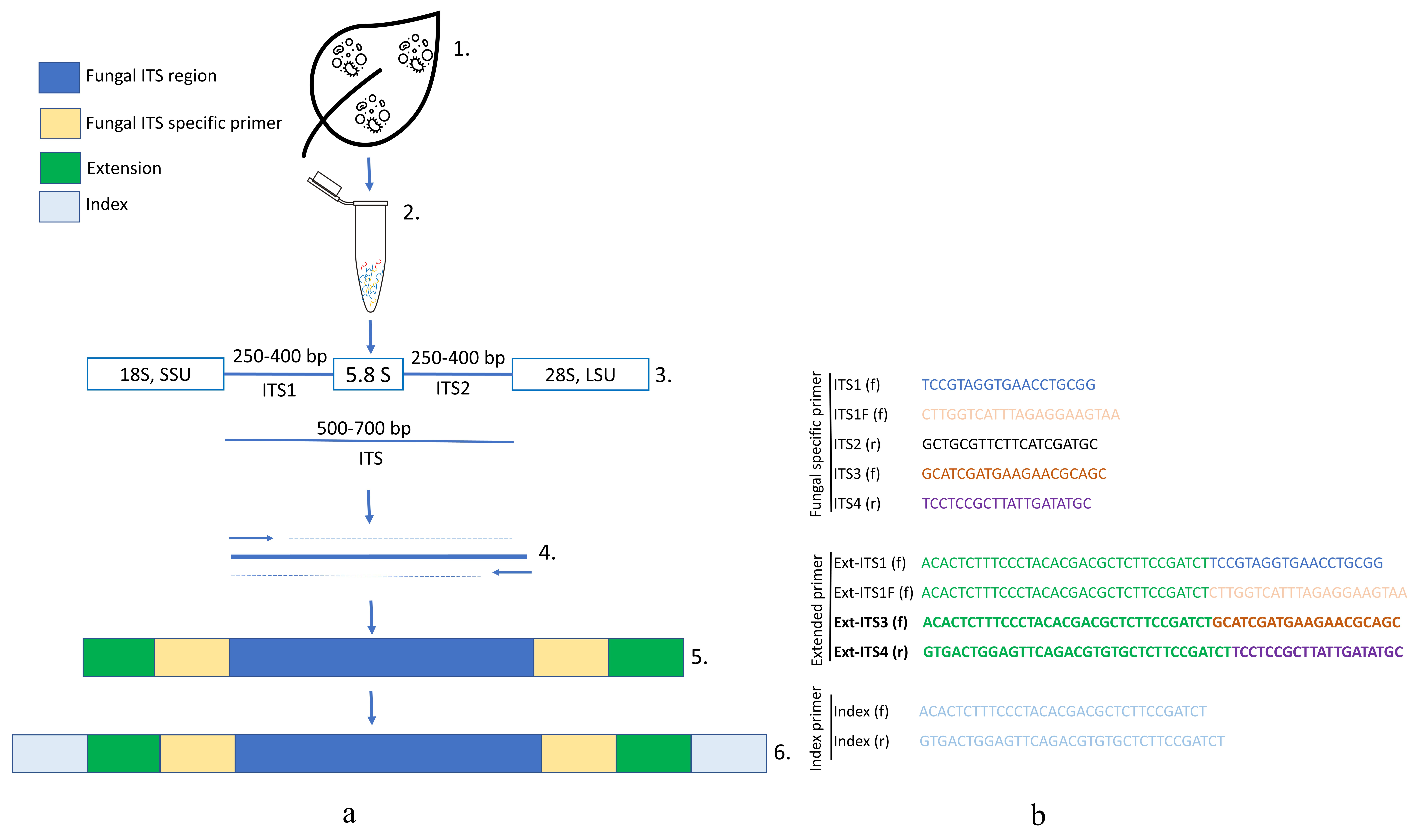


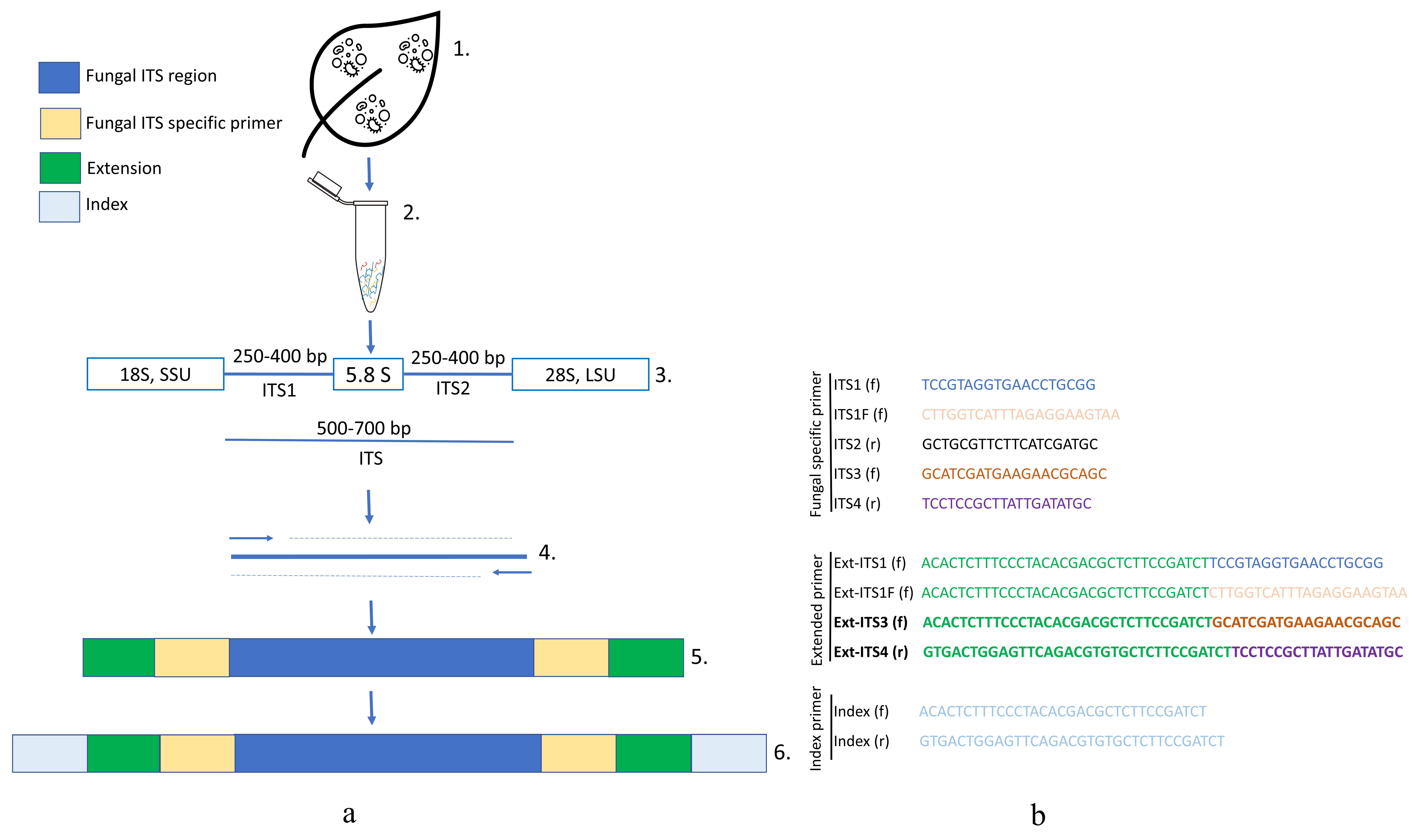{kind=link}
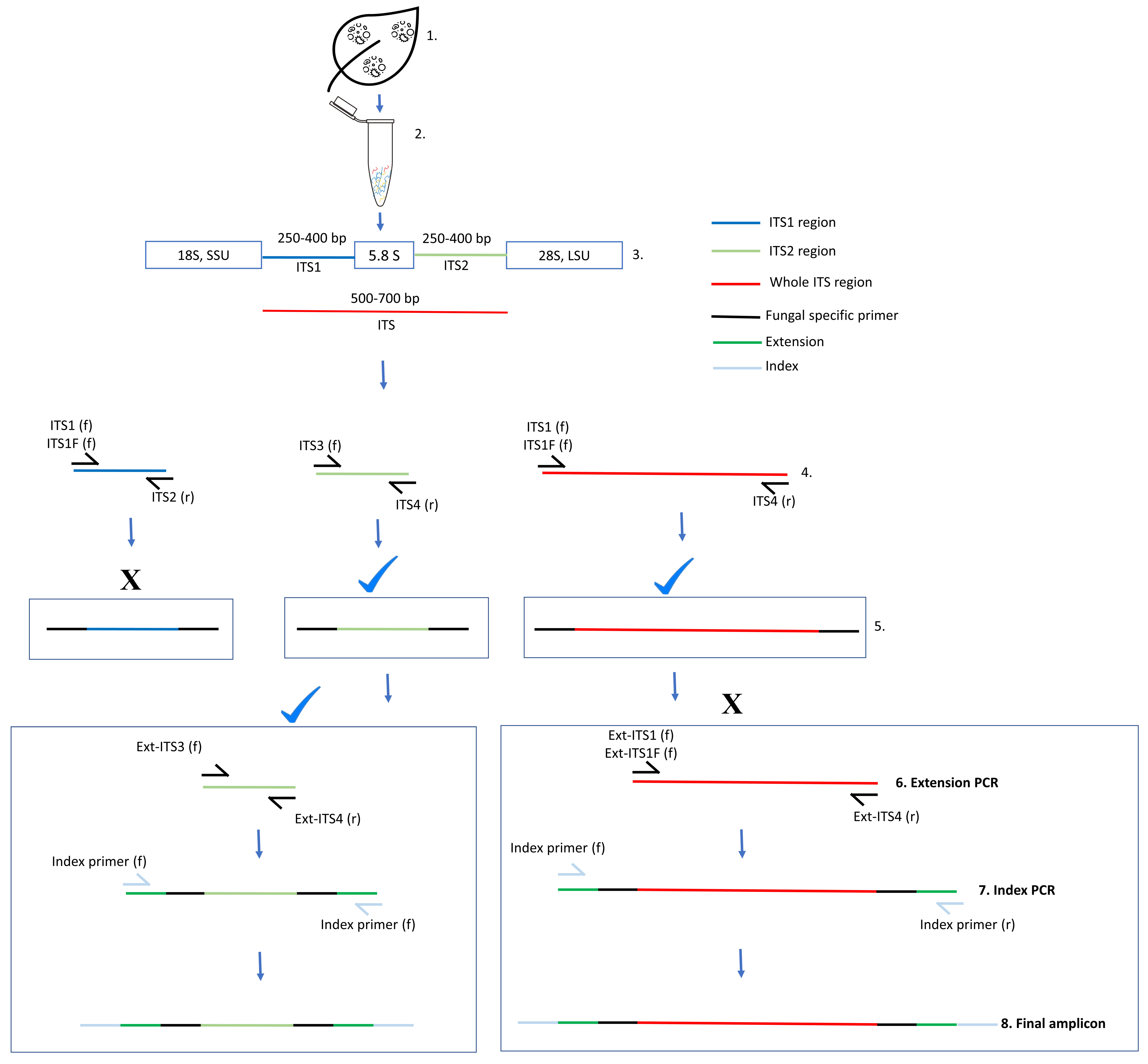
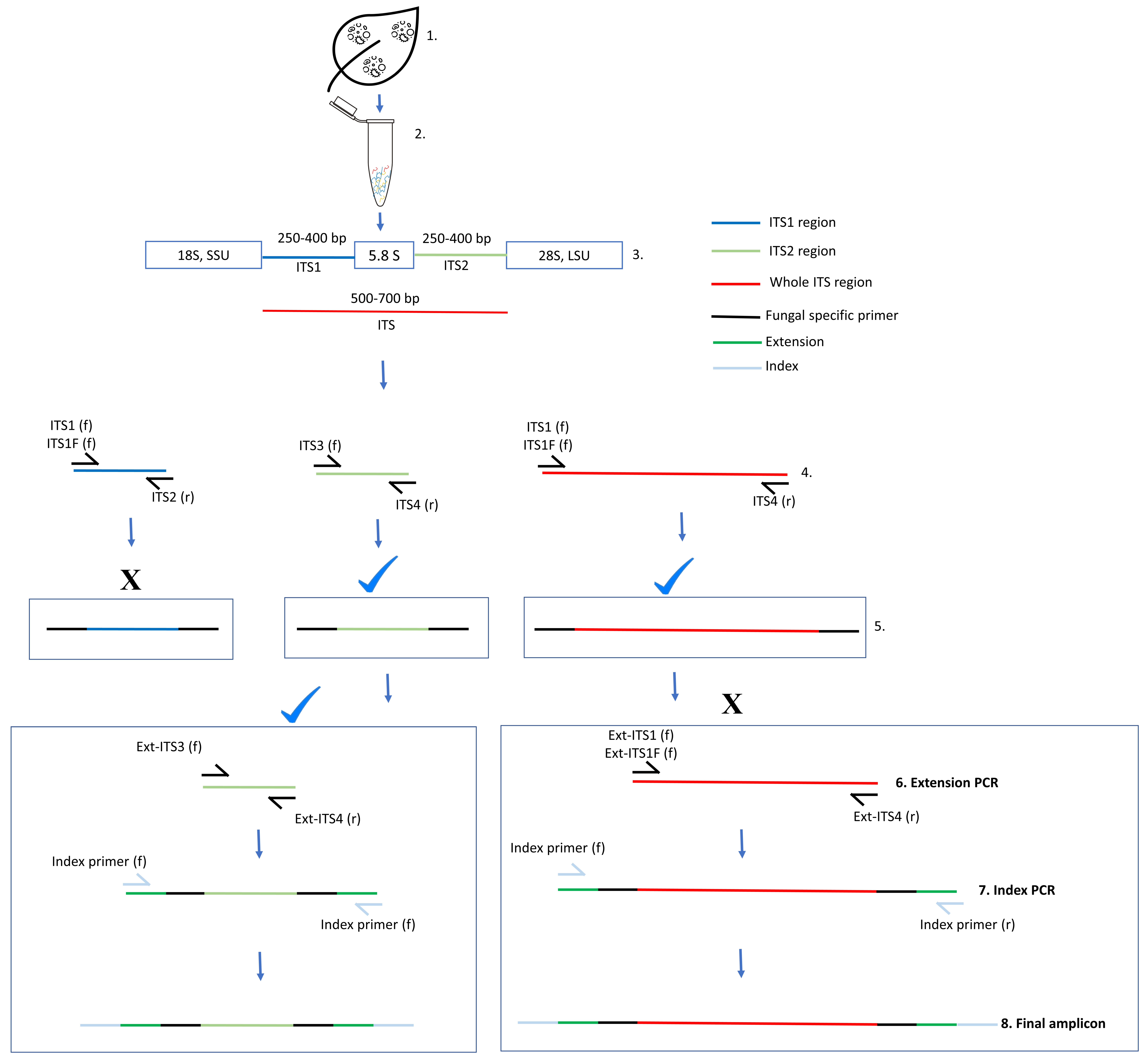{kind=link}
{kind=link}
| Kit | Sample ID | DNA Concentration (ng/µL) | 260/280 | 260/230 |
|---|---|---|---|---|
| E.Z.N.A. Plant DNA | 253 | 561.1 | 2.04 | 1.99 |
| 281 | 804.3 | 2.02 | 2.00 | |
| ChargeSwitch gDNA | 253 | 1526.3 | 0.63 | 0.29 |
| 281 | 1311.8 | 0.63 | 0.26 |
| Purpose | Primers | Template DNA | Settings | Outcome | Decision/Selection |
|---|---|---|---|---|---|
| Fungal PCR for ITS-selection | ITS1 (f) + ITS4 (r) | 1. 1 µL | 1. 56/30 * | 1. SB with desired DF (a) | 1. Pp failed to bind to fungal ITS region. |
| ITS1F (f) + ITS4 (r) | 1. 1 µL | 1. 56/30 2. 57/30 | 1. NB 2. SB with desired DF (b) | 1. Pp failed to bind to ITS. 2. Sucessful amplification of ITS. | |
| ITS1 (f) + ITS2 (r) | 1. 1 µL | 1. 56/30 2. 57/30 | 1. NB 2. NB | 1. Pp failed to bind to ITS1. 2. Pp failed to bind to ITS1. | |
| ITS3 (f) + ITS4 (r) | 1. 1 µL | 1. 57/30 | 1. SB with desired DF (c) | 1. Pp successfully amplified ITS2. | |
| Ext. PCR for ITS ampl. | Ext-ITS1F (f) + Ext-ITS4 (r) | 1. 1 µL 2. 1 µL 3. 1 µL 4. 1 and 0.5 µL 5. 1 and 0.5 µL 6. 1 and 0.5 µL 7. 1 and 0.5 µL 8. 0.5 and 1 µL PCR Product 9. 1 and 0.5 µL | 1. 62/30 2. 60/30 3. 58/30 4. 62/30 5. 64/30 6. 66/30 7. 68/30 8. 60/30 9. 58/33 | 1. FB (d) 2. FB with PD 3. FB with undesired DF (e) 4. FB with undesired DF 5. FB with undesired DF 6. FB with undesired DF 7. FB with undesired DF 8. SB with desired DF 9. PD with both desired and undesired DF (f) | 1–7. Ext. Pp failed to amplify the ITS. 8. Ext. Pp successfully amplified ITS 9. Ext. Pp amplified successfully. |
| Ext-ITS1 (f) + Ext-ITS4 (r) | 1. 1 µL 2. 1 µL | 1. 56/30 2. 58/30 | 1. NB 2. NB | 1. Ext. Pp failed to amplify ITS. 2. Ext. Pp failed to amplify ITS. | |
| Index PCR | Index primer pair | 1. 8 µL of PCR product of Ext-ITS1F (f)+ Ext-ITS4 (r) | 1. 55/8 | 1. FB with PD (h) | 1. Index Pp failed to amplify, suggesting absence of ext. region in PCR products. |
| Ext. PCR | Ext-ITS3 (f) + Ext-ITS4 (r) | 1. 1 µL 2. 1 µL 3. 1 µL | 1. 72/33 2. 70/33 3. 57/33 | 1. NB (g) 2. FB (g) 3. SB with desired DF (g) | 1. Ext Pp failed to amplify ITS2. 2. Ext Pp failed to amplify ITS2. 3. Ext Pp amplified ITS2. |
| Index PCR for ITS2 ampl. | Index primers | 1. 8 µL PCR product of Ext-ITS3 (f) + Ext-ITS4 (r) | 1. 55/8 | 1. Desired difference between index PCR and Ext. PCR(i). | 1.Index Pp successfully amplified extended overhang with index for amplicon library. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddique, A.B.; Albrectsen, B.R.; Ilbi, H.; Siddique, A.B. Optimization of Protocol for Construction of Fungal ITS Amplicon Library for High-Throughput Illumina Sequencing to Study the Mycobiome of Aspen Leaves. Appl. Sci. 2022, 12, 1136. https://doi.org/10.3390/app12031136
Siddique AB, Albrectsen BR, Ilbi H, Siddique AB. Optimization of Protocol for Construction of Fungal ITS Amplicon Library for High-Throughput Illumina Sequencing to Study the Mycobiome of Aspen Leaves. Applied Sciences. 2022; 12(3):1136. https://doi.org/10.3390/app12031136
Chicago/Turabian StyleSiddique, Abu Bakar, Benedicte Riber Albrectsen, Hulya Ilbi, and Abu Bakar Siddique. 2022. "Optimization of Protocol for Construction of Fungal ITS Amplicon Library for High-Throughput Illumina Sequencing to Study the Mycobiome of Aspen Leaves" Applied Sciences 12, no. 3: 1136. https://doi.org/10.3390/app12031136
APA StyleSiddique, A. B., Albrectsen, B. R., Ilbi, H., & Siddique, A. B. (2022). Optimization of Protocol for Construction of Fungal ITS Amplicon Library for High-Throughput Illumina Sequencing to Study the Mycobiome of Aspen Leaves. Applied Sciences, 12(3), 1136. https://doi.org/10.3390/app12031136