
Carbazole Derivatives as STAT Inhibitors: An Overview


 ,
, 
 and
and 
Abstract
1. Introduction
2. STAT Proteins: Novel Molecular Targets for Cancer Drug Discovery
3. STATs Inhibitors
3.1. Peptides and Small Molecules as STATs Inhibitors
3.2. Natural Compounds as STATs Inhibitors
3.3. Carbazoles as STAT Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chin, Y.W.; Balunas, M.J.; Chai, H.B.; Kinghorn, A.D. Drug discovery from natural sources. AAPS J. 2006, 8, E239–E253. [Google Scholar] [CrossRef]
- Cordell, G.A.; Quinn-Beattie, M.L.; Farnsworth, N.R. The potential of alkaloids in drug discovery. Phytother Res. 2001, 15, 183–205. [Google Scholar] [CrossRef]
- Bhambhani, S.; Kondhare, K.R.; Giri, A.P. Diversity in chemical structures and biological properties of plant alkaloids. Molecules 2021, 26, 3374. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Mital, A. Synthetic nitroimidazoles: Biological activities and mutagenicity relationships. Sci. Pharm. 2009, 77, 497–520. [Google Scholar] [CrossRef]
- Nekrasov, D. Biological activity of 5-and 6-membered azaheterocycles and their synthesis from 5-aryl-2, 3-dihydrofuran-2, 3-diones. Chem. Heterocycl. Compd. 2001, 37, 263–275. [Google Scholar] [CrossRef]
- Laurence, C.; Brameld, K.A.; Graton, J.; Le Questel, J.-Y.; Renault, E. The p K BHX database: Toward a better understanding of hydrogen-bond basicity for medicinal chemists. J. Med. Chem. 2009, 52, 4073–4086. [Google Scholar] [CrossRef] [PubMed]
- Graebe, C.; Glaser, C. Ueber carbazol. Justus Liebigs Ann. Chem. 1872, 163, 343–360. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Barman, B.K.; Bose, P.K. On the constitution of murrayanine, a carbazole derivative isolated from Murraya koenigii Spreng. Tetrahedron 1965, 21, 681–685. [Google Scholar] [CrossRef]
- Saturnino, C.; Caruso, A.; Longo, P.; Capasso, A.; Pingitore, A.; Cristina Caroleo, M.; Cione, E.; Perri, M.; Nicolò, F.; Mollica Nardo, V. Crystallographic study and biological evaluation of 1, 4-dimethyl-N-alkylcarbazoles. Curr. Top. Med. Chem. 2015, 15, 973–979. [Google Scholar] [CrossRef]
- Panno, A.; Sinicropi, M.S.; Caruso, A.; El-Kashef, H.; Lancelot, J.C.; Aubert, G.; Lesnard, A.; Cresteil, T.; Rault, S. New trimethoxybenzamides and trimethoxyphenylureas derived from dimethylcarbazole as cytotoxic agents. Part, I. J. Heterocycl. Chem. 2014, 51, E294–E302. [Google Scholar] [CrossRef]
- Caruso, A.; Sinicropi, M.S.; Lancelot, J.-C.; El-Kashef, H.; Saturnino, C.; Aubert, G.; Ballandonne, C.; Lesnard, A.; Cresteil, T.; Dallemagne, P. Synthesis and evaluation of cytotoxic activities of new guanidines derived from carbazoles. Bioorg. Med. Chem. Lett. 2014, 24, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Sinicropi, M.S.; Iacopetta, D.; Rosano, C.; Randino, R.; Caruso, A.; Saturnino, C.; Muià, N.; Ceramella, J.; Puoci, F.; Rodriquez, M. N-thioalkylcarbazoles derivatives as new anti-proliferative agents: Synthesis, characterisation and molecular mechanism evaluation. J. Enzyme Inhib. Med. Chem. 2018, 33, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Knölker, H.-J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4428. [Google Scholar] [CrossRef]
- Bashir, M.; Bano, A.; Ijaz, A.S.; Chaudhary, B.A. Recent developments and biological activities of N-substituted carbazole derivatives: A review. Molecules 2015, 20, 13496–13517. [Google Scholar] [CrossRef] [PubMed]
- Is, O.D.; Koyuncu, F.B.; Koyuncu, S.; Ozdemir, E. A new imine coupled pyrrole–carbazole–pyrrole polymer: Electro-optical properties and electrochromism. Polymer 2010, 51, 1663–1669. [Google Scholar] [CrossRef]
- Srinivas, K.; Kumar, C.R.; Reddy, M.A.; Bhanuprakash, K.; Rao, V.J.; Giribabu, L. D-π-A organic dyes with carbazole as donor for dye-sensitized solar cells. Synth. Met. 2011, 161, 96–105. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, D.U.; Choo, D.C.; Kim, T.W.; Ryu, E.D.; Kim, S.W.; Lim, J.S. Organic light-emitting devices fabricated utilizing core/shell CdSe/ZnS quantum dots embedded in a polyvinylcarbazole. J. Mater. Sci. 2011, 46, 1239–1243. [Google Scholar] [CrossRef]
- Wang, L.; Fu, Y.; Zhu, L.; Cui, G.; Liang, F.; Guo, L.; Zhang, X.; Xie, Z.; Su, Z. Synthesis and photovoltaic properties of low-bandgap polymers based on N-arylcarbazole. Polymer 2011, 52, 1748–1754. [Google Scholar] [CrossRef]
- Adhikari, R.M.; Neckers, D.C.; Shah, B.K. Photophysical study of blue, green, and orange-red light-emitting carbazoles. J. Org. Chem. 2009, 74, 3341–3349. [Google Scholar] [CrossRef]
- Caruso, A.; Ceramella, J.; Iacopetta, D.; Saturnino, C.; Mauro, M.V.; Bruno, R.; Aquaro, S.; Sinicropi, M.S. Carbazole derivatives as antiviral agents: An overview. Molecules 2019, 24, 1912. [Google Scholar] [CrossRef]
- Caruso, A.; Iacopetta, D.; Puoci, F.; Rita Cappello, A.; Saturnino, C.; Stefania Sinicropi, M. Carbazole derivatives: A promising scenario for breast cancer treatment. Mini Rev. Med. Chem 2016, 16, 630–643. [Google Scholar] [CrossRef]
- Roy, J.; Jana, A.K.; Mal, D. Recent trends in the synthesis of carbazoles: An update. Tetrahedron 2012, 68, 6099–6121. [Google Scholar] [CrossRef]
- Caruso, A.; Voisin-Chiret, A.S.; Lancelot, J.-C.; Sinicropi, M.S.; Garofalo, A.; Rault, S. Novel and efficient synthesis of 5, 8-dimethyl-9H-carbazol-3-ol via a hydroxydeboronation reaction. Heterocycles 2007, 71, 2203–2210. [Google Scholar] [CrossRef]
- Caruso, A.; Voisin-Chiret, A.S.; Lancelot, J.-C.; Sinicropi, M.S.; Garofalo, A.; Rault, S. Efficient and simple synthesis of 6-Aryl-1, 4-dimethyl-9H-carbazoles. Molecules 2008, 13, 1312–1320. [Google Scholar] [CrossRef]
- Saturnino, C.; Grande, F.; Aquaro, S.; Caruso, A.; Iacopetta, D.; Bonomo, M.G.; Longo, P.; Schols, D.; Sinicropi, M.S. Chloro-1, 4-dimethyl-9H-carbazole derivatives displaying anti-HIV activity. Molecules 2018, 23, 286. [Google Scholar] [CrossRef]
- You, X.; Zhu, D.; Lu, W.; Sun, Y.; Qiao, S.; Luo, B.; Du, Y.; Pi, R.; Hu, Y.; Huang, P. Design, synthesis and biological evaluation of N-arylsulfonyl carbazoles as novel anticancer agents. RSC Adv. 2018, 8, 17183–17190. [Google Scholar] [CrossRef]
- Ceramella, J.; Iacopetta, D.; Barbarossa, A.; Caruso, A.; Grande, F.; Bonomo, M.G.; Mariconda, A.; Longo, P.; Carmela, S.; Sinicropi, M.S. Carbazole derivatives as kinase-targeting inhibitors for cancer treatment. Mini Rev. Med. Chem 2020, 20, 444–465. [Google Scholar] [CrossRef] [PubMed]
- Ceramella, J.; Caruso, A.; Occhiuzzi, M.A.; Iacopetta, D.; Barbarossa, A.; Rizzuti, B.; Dallemagne, P.; Rault, S.; El-Kashef, H.; Saturnino, C. Benzothienoquinazolinones as new multi-target scaffolds: Dual inhibition of human Topoisomerase I and tubulin polymerization. Eur. J. Med. Chem. 2019, 181, 111583. [Google Scholar] [CrossRef] [PubMed]
- Saturnino, C.; Caruso, A.; Iacopetta, D.; Rosano, C.; Ceramella, J.; Muià, N.; Mariconda, A.; Bonomo, M.G.; Ponassi, M.; Rosace, G. Inhibition of human topoisomerase II by N, N, N-trimethylethanammonium iodide alkylcarbazole derivatives. Chem. Med. Chem. 2018, 13, 2635–2643. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Govindarajan, B.; Battle, T.E.; Lynch, R.; Frank, D.A.; Ushio-Fukai, M.; Perry, B.N.; Stern, D.F.; Bowden, G.T.; Liu, A. Carbazole is a naturally occurring inhibitor of angiogenesis and inflammation isolated from antipsoriatic coal tar. J. Invest. Dermatol. 2006, 126, 1396–1402. [Google Scholar] [CrossRef]
- Saturnino, C.; Palladino, C.; Napoli, M.; Sinicropi, M.S.; Botta, A.; Sala, M.; de Prati, A.C.; Novellino, E.; Suzuki, H. Synthesis and biological evaluation of new N-alkylcarbazole derivatives as STAT3 inhibitors: Preliminary study. Eur. J. Med. Chem. 2013, 60, 112–119. [Google Scholar] [CrossRef]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef]
- Smithgall, T.E.; Briggs, S.D.; Schreiner, S.; Lerner, E.C.; Cheng, H.; Wilson, M.B. Control of myeloid differentiation and survival by Stats. Oncogene 2000, 19, 2612–2618. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Gopal, R.; Manni, M.L.; McHugh, K.J.; Mandalapu, S.; Robinson, K.M.; Alcorn, J.F. STAT1 is required for suppression of type 17 immunity during influenza and bacterial superinfection. Immuno. Horizons 2017, 1, 81–91. [Google Scholar] [CrossRef]
- Machiraju, D.; Moll, I.; Gebhardt, C.; Sucker, A.; Buder-Bakhaya, K.; Schadendorf, D.; Hassel, J.C. STAT5 expression correlates with recurrence and survival in melanoma patients treated with interferon-α. Melanoma Res. 2018, 28, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Gamero, A.M.; Young, M.R.; Mentor-Marcel, R.; Bobe, G.; Scarzello, A.J.; Wise, J.; Colburn, N.H. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev. Res. 2010, 3, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Akira, S. Functional roles of STAT family proteins: Lessons from knockout mice. Stem Cells. 1999, 17, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Macias, E.; Rao, D.; DiGiovanni, J. Role of stat3 in skin carcinogenesis: Insights gained from relevant mouse models. J. Skin Cancer 2013, 2013, 684050. [Google Scholar] [CrossRef]
- Darnell, J.E. Validating stat3 in cancer therapy. Nat. Med. 2005, 11, 595–596. [Google Scholar] [CrossRef]
- Tweardy, D.J.; Jing, N. Enhancing or eliminating signals for cell survival to treat disease. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 33. [Google Scholar]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar]
- Gao, P.; Niu, N.; Wei, T.; Tozawa, H.; Chen, X.; Zhang, C.; Zhang, J.; Wada, Y.; Kapron, C.M.; Liu, J. The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget 2017, 8, 69139–69161. [Google Scholar] [CrossRef]
- Dutta, P.; Zhang, L.; Zhang, H.; Peng, Q.; Montgrain, P.R.; Wang, Y.; Song, Y.; Li, J.; Li, W.X. Unphosphorylated STAT3 in heterochromatin formation and tumor suppression in lung cancer. BMC Cancer 2020, 20, 145. [Google Scholar] [CrossRef]
- Loh, C.Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: Functions and therapeutic implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef]
- Andersson, E.I.; Brück, O.; Braun, T.; Mannisto, S.; Saikko, L.; Lagström, S.; Ellonen, P.; Leppä, S.; Herling, M.; Kovanen, P.E.; et al. STAT3 mutation is associated with STAT3 activation in CD30+ ALK− ALCL. Cancers 2020, 12, 702. [Google Scholar] [CrossRef]
- Szabo, S.J.; Sullivan, B.M.; Peng, S.L.; Glimcher, L.H. Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 2003, 21, 713–758. [Google Scholar] [CrossRef]
- Lovett-Racke, A.E.; Yang, Y.; Racke, M.K. Th1 versus Th17: Are T cell cytokines relevant in multiple sclerosis? Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 246–251. [Google Scholar] [CrossRef]
- Lin, J.-X.; Leonard, W.J. The role of stat5a and stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576. [Google Scholar] [CrossRef]
- Buitenhuis, M.; Baltus, B.; Lammers, J.-W.J.; Coffer, P.J.; Koenderman, L. Signal transducer and activator of transcription 5a (STAT5a) is required for eosinophil differentiation of human cord blood–derived CD34+ cells. Blood 2003, 101, 134–142. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef]
- Cotarla, I.; Ren, S.; Zhang, Y.; Gehan, E.; Singh, B.; Furth, P.A. Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. Int. J. Cancer 2004, 108, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Benekli, M.; Baumann, H.; Wetzler, M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 2009, 27, 4422. [Google Scholar] [CrossRef]
- Nelson, E.A.; Sharma, S.V.; Settleman, J.; Frank, D.A. A chemical biology approach to developing STAT inhibitors: Molecular strategies for accelerating clinical translation. Oncotarget 2011, 2, 518. [Google Scholar] [CrossRef] [PubMed]
- Kaymaz, B.T.; Selvi, N.; Gokbulut, A.A.; Aktan, C.; Gündüz, C.; Saydam, G.; Sahin, F.; Cetintaş, V.B.; Baran, Y.; Kosova, B. Suppression of STAT5A and STAT5B chronic myeloid leukemia cells via siRNA and antisense-oligonucleotide applications with the induction of apoptosis. Am. J. Blood Res. 2013, 3, 58–70. [Google Scholar]
- Sehra, S.; Yao, Y.; Howell, M.D.; Nguyen, E.T.; Kansas, G.S.; Leung, D.Y.; Travers, J.B.; Kaplan, M.H. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J. Immunol. 2010, 184, 3186–3190. [Google Scholar] [CrossRef]
- Chapoval, S.P.; Dasgupta, P.; Smith, E.P.; DeTolla, L.J.; Lipsky, M.M.; Kelly-Welch, A.E.; Keegan, A.D. STAT6 expression in multiple cell types mediates the cooperative development of allergic airway disease. J. Immunol. 2011, 186, 2571–2583. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Nakazawa, S.; Todoroki, M.; Shinozaki, K.; Sakai, H.; Misawa, M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am. J. Respir. Cell Mol. Biol. 2009, 40, 159–167. [Google Scholar] [CrossRef]
- Chiba, Y.; Todoroki, M.; Nishida, Y.; Tanabe, M.; Misawa, M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 516–524. [Google Scholar] [CrossRef]
- Khaled, W.T.; Read, E.K.; Nicholson, S.E.; Baxter, F.O.; Brennan, A.J.; Came, P.J.; Sprigg, N.; McKenzie, A.N.; Watson, C.J. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development 2007, 134, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, C.; Mao, H.; Li, Z.; Fang, Z.; Chen, Q.; Lin, M.; Jiang, X.; Hu, Y.; Wang, W.; et al. E2F1/SP3/STAT6 axis is required for IL-4-induced epithelial-mesenchymal transition of colorectal cancer cells. Int. J. Oncol. 2018, 53, 567–578. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Sarid, N.; Eshel, R.; Rahamim, E.; Carmiel, M.; Kirgner, I.; Shpringer, M.; Trestman, S.; Marilus, R.; Perry, C.; Polliack, A.; et al. JAK2 mutation: An aid in the diagnosis of occult myeloproliferative neoplasms in patients with major intraabdominal vein thrombosis and normal blood counts. Isr. Med. Assoc. J. 2013, 15, 698–700. [Google Scholar] [PubMed]
- López-Matencio, J.M.S.; Baladrón, A.M.; Castañeda, S. JAK-STAT inhibitors for the treatment of immunomediated diseases. Med. Clin. 2019, 152, 353–360. [Google Scholar] [CrossRef]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; De Groot, R.P.; Jove, R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Zong, C.S.; Zeng, L.; Jiang, Y.; Sadowski, H.B.; Wang, L.-H. Stat3 plays an important role in oncogenic Ros-and insulin-like growth factor I receptor-induced anchorage-independent growth. J. Biol. Chem. 1998, 273, 28065–28072. [Google Scholar] [CrossRef]
- Zhang, N.; Zheng, Q.; Wang, Y.; Lin, J.; Wang, H.; Liu, R.; Yan, M.; Chen, X.; Yang, J.; Chen, X. Renoprotective effect of the recombinant anti-IL-6R fusion proteins by inhibiting JAK2/STAT3 signaling pathway in diabetic nephropathy. Front. Pharmacol. 2021, 12, 681424. [Google Scholar] [CrossRef]
- Wen, X.; Lin, H.H.; Shih, H.-M.; Kung, H.-J.; Ann, D.K. Kinase activation of the non-receptor tyrosine kinase Etk/BMX alone is sufficient to transactivate STAT-mediated gene expression in salivary and lung epithelial cells. J. Biol. Chem. 1999, 274, 38204–38210. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.; Medveczky, M.M.; Medveczky, P.G. Herpesvirus saimiri Tip-484 membrane protein markedly increases p56lck activity in T cells. J. Virol. 1997, 71, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Migone, T.S.; Lin, J.X.; Cereseto, A.; Mulloy, J.C.; O’Shea, J.J.; Franchini, G.; Leonard, W.J. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 1995, 269, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Yu, C.L.; Hudnall, A.; Catlett, R.; Nelson, K.L.; Smithgall, T.; Fujita, D.J.; Ethier, S.P.; Jove, R. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997, 8, 1267–1276. [Google Scholar] [PubMed]
- Weber-Nordt, R.; Egen, C.; Wehinger, J.; Ludwig, W.; Gouilleux-Gruart, V.; Mertelsmann, R.; Finke, J. Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood 1996, 88, 809–816. [Google Scholar] [CrossRef]
- Gharibi, T.; Babaloo, Z.; Hosseini, A.; Abdollahpour-alitappeh, M.; Hashemi, V.; Marofi, F.; Nejati, K.; Baradaran, B. Targeting STAT3 in cancer and autoimmune diseases. Eur. J. Pharmacol. 2020, 878, 173107. [Google Scholar] [CrossRef]
- Turkson, J. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets 2004, 8, 409–422. [Google Scholar] [CrossRef]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.S.; Ren, Z.; Mandal, P.K.; Chen, X. Model of intermolecular interactions between high affinity phosphopeptides and Stat3. Adv. Exp. Med. Biol. 2009, 611, 543–544. [Google Scholar]
- McMurray, J.S. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers 2008, 90, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Drewry, J.A.; Shahani, V.M.; Page, B.D.; Gunning, P.T. Molecular disruption of oncogenic signal transducer and activator of transcription 3 (STAT3) protein. Biochem. Cell Biol. 2009, 87, 825–833. [Google Scholar] [CrossRef]
- Zhao, W.; Jaganathan, S.; Turkson, J. A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro. J. Biol. Chem. 2010, 285, 35855–35865. [Google Scholar] [CrossRef]
- McMurray, J.S. A new small-molecule Stat3 inhibitor. Chem. Biol. 2006, 13, 1123–1124. [Google Scholar] [CrossRef] [PubMed]
- Fuh, B.; Sobo, M.; Cen, L.; Josiah, D.; Hutzen, B.; Cisek, K.; Bhasin, D.; Regan, N.; Lin, L.; Chan, C. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 2009, 100, 106–112. [Google Scholar] [CrossRef]
- Hao, W.; Hu, Y.; Niu, C.; Huang, X.; Chang, C.-P.B.; Gibbons, J.; Xu, J. Discovery of the catechol structural moiety as a Stat3 SH2 domain inhibitor by virtual screening. Bioorg. Med. Chem. Lett. 2008, 18, 4988–4992. [Google Scholar] [CrossRef]
- Siddiquee, K.A.; Gunning, P.T.; Glenn, M.; Katt, W.P.; Zhang, S.; Schroeck, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; et al. Identification of a New Series of STAT3 Inhibitors by Virtual Screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013, 62, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Nam, H.J.; Kim, H.P.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Liu, Y.Y.; Ma, Y.G.; Xue, Y.X.; Liu, D.G.; Ren, Y.; Liu, X.B.; Li, Y.; Li, Z. Curcumin blocks small cell lung cancer cells migration, invasion, angiogenesis, cell cycle and neoplasia through Janus kinase-STAT3 signalling pathway. PLoS ONE 2012, 7, e37960. [Google Scholar] [CrossRef]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.-K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 2011, 11, 1–15. [Google Scholar] [CrossRef]
- Selvendiran, K.; Ahmed, S.; Dayton, A.; Kuppusamy, M.L.; Rivera, B.K.; Kálai, T.; Hideg, K.; Kuppusamy, P. HO-3867, a curcumin analog, sensitizes cisplatin-resistant ovarian carcinoma, leading to therapeutic synergy through STAT3 inhibition. Cancer Biol. Ther. 2011, 12, 837–845. [Google Scholar] [CrossRef]
- Tierney, B.J.; McCann, G.A.; Cohn, D.E.; Eisenhauer, E.; Sudhakar, M.; Kuppusamy, P.; Hideg, K.; Selvendiran, K. HO-3867, a STAT3 inhibitor induces apoptosis by inactivation of STAT3 activity in BRCA1-mutated ovarian cancer cells. Cancer Biol. Ther. 2012, 13, 766–775. [Google Scholar] [CrossRef]
- Onimoe, G.I.; Liu, A.; Lin, L.; Wei, C.C.; Schwartz, E.B.; Bhasin, D.; Li, C.; Fuchs, J.R.; Li, P.K.; Houghton, P.; et al. Small molecules, LLL12 and FLLL32, inhibit STAT3 and exhibit potent growth suppressive activity in osteosarcoma cells and tumor growth in mice. Invest. New Drugs 2012, 30, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Bid, H.K.; Oswald, D.; Li, C.; London, C.A.; Lin, J.; Houghton, P.J. Anti-angiogenic activity of a small molecule STAT3 inhibitor LLL12. PLoS ONE 2012, 7, e35513. [Google Scholar] [CrossRef]
- Shakibaei, M.; Harikumar, K.B.; Aggarwal, B.B. Resveratrol addiction: To die or not to die. Mol. Nutr. Food Res. 2009, 53, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, H.S.; Shin, Y.-J.; Lee, C.S.; Won, C.; Lee, S.-A.; Lee, J.W.; Kim, Y.; Kang, J.-S.; Ye, S.-K. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp. Mol. Med. 2008, 40, 514–522. [Google Scholar] [CrossRef]
- Sarkar, S.; Dutta, D.; Samanta, S.K.; Bhattacharya, K.; Pal, B.C.; Li, J.; Datta, K.; Mandal, C.; Mandal, C. Oxidative inhibition of Hsp90 disrupts the super-chaperone complex and attenuates pancreatic adenocarcinoma in vitro and in vivo. Int. J. Cancer 2013, 132, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Itoigawa, M.; Nakao, K.; Murata, T.; Tsuboi, M.; Kaneda, N.; Furukawa, H. Induction of apoptosis by carbazole alkaloids isolated from Murraya koenigii. Phytomedicine 2006, 13, 359–365. [Google Scholar] [CrossRef]
- Roy, M.K.; Thalang, V.N.; Trakoontivakorn, G.; Nakahara, K. Mahanine, a carbazole alkaloid from Micromelum minutum, inhibits cell growth and induces apoptosis in U937 cells through a mitochondrial dependent pathway. Br. J. Pharmacol. 2005, 145, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.K.; Thalang, V.N.; Trakoontivakorn, G.; Nakahara, K. Mechanism of mahanine-induced apoptosis in human leukemia cells (HL-60). Biochem. Pharmacol. 2004, 67, 41–51. [Google Scholar] [CrossRef]
- Sinha, S.; Pal, B.C.; Jagadeesh, S.; Banerjee, P.P.; Bandyopadhaya, A.; Bhattacharya, S. Mahanine inhibits growth and induces apoptosis in prostate cancer cells through the deactivation of Akt and activation of caspases. Prostate 2006, 66, 1257–1265. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Söti, C.; Csermely, P. Inhibition of Hsp90: A new strategy for inhibiting protein kinases. Biochim. Biophys. Acta Proteins Proteom. 2004, 1697, 233–242. [Google Scholar] [CrossRef]
- Saturnino, C.; Iacopetta, D.; Sinicropi, M.S.; Rosano, C.; Caruso, A.; Caporale, A.; Marra, N.; Marengo, B.; Pronzato, M.A.; Parisi, O.I. N-alkyl carbazole derivatives as new tools for Alzheimer’s disease: Preliminary studies. Molecules 2014, 19, 9307–9317. [Google Scholar] [CrossRef]
- Hou, S.; Yi, Y.W.; Kang, H.J.; Zhang, L.; Kim, H.J.; Kong, Y.; Liu, Y.; Wang, K.; Kong, H.-S.; Grindrod, S. Novel carbazole inhibits phospho-STAT3 through induction of protein–tyrosine phosphatase PTPN6. J. Med. Chem. 2014, 57, 6342–6353. [Google Scholar] [CrossRef]
- Kong, Y.; Jung, M.; Wang, K.; Grindrod, S.; Velena, A.; Lee, S.A.; Dakshanamurthy, S.; Yang, Y.; Miessau, M.; Zheng, C. Histone deacetylase cytoplasmic trapping by a novel fluorescent HDAC inhibitor. Mol. Cancer Ther. 2011, 10, 1591–1599. [Google Scholar] [CrossRef]
- Kong, H.-S.; Tian, S.; Kong, Y.; Du, G.; Zhang, L.; Jung, M.; Dritschilo, A.; Brown, M.L. Preclinical studies of YK-4-272, an inhibitor of class II histone deacetylases by disruption of nucleocytoplasmic shuttling. Pharm. Res. 2012, 29, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Walls, T.H.; Grindrod, S.C.; Beraud, D.; Zhang, L.; Baheti, A.R.; Dakshanamurthy, S.; Patel, M.K.; Brown, M.L.; MacArthur, L.H. Synthesis and biological evaluation of a fluorescent analog of phenytoin as a potential inhibitor of neuropathic pain and imaging agent. Bioorg. Med. Chem. 2012, 20, 5269–5276. [Google Scholar] [CrossRef][Green Version]
- Kang, H.J.; Yi, Y.W.; Hou, S.-J.; Kim, H.J.; Kong, Y.; Bae, I.; Brown, M.L. Disruption of STAT3-DNMT1 interaction by SH-I-14 induces re-expression of tumor suppressor genes and inhibits growth of triple-negative breast tumor. Oncotarget 2017, 8, 83457. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3-and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef]
- Cuenca-López, M.D.; Serrano-Heras, G.; Montero, J.C.; Corrales-Sánchez, V.; Gomez-Juarez, M.; Gascón-Escribano, M.J.; Morales, J.C.; Voisin, V.; Núñez, L.E.; Morís, F. Antitumor activity of the novel multi-kinase inhibitor EC-70124 in triple negative breast cancer. Oncotarget 2015, 6, 27923. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Estupiñan, O.; Santos, L.; Rodriguez, A.; Fernandez-Nevado, L.; Costales, P.; Perez-Escuredo, J.; Hermosilla, M.A.; Oro, P.; Rey, V.; Tornin, J. The multikinase inhibitor EC-70124 synergistically increased the antitumor activity of doxorubicin in sarcomas. Int. J. Cancer 2019, 145, 254–266. [Google Scholar] [CrossRef]
- Botta, A.; Sirignano, E.; Popolo, A.; Saturnino, C.; Terracciano, S.; Foglia, A.; Sinicropi, M.S.; Longo, P.; Di Micco, S. Identification of lead compounds as inhibitors of STAT3: Design, synthesis and bioactivity. Mol. Inform. 2015, 34, 689–697. [Google Scholar] [CrossRef]
- Zimmermann, K.; Sang, X.; Mastalerz, H.A.; Johnson, W.L.; Zhang, G.; Liu, Q.; Batt, D.; Lombardo, L.J.; Vyas, D.; Trainor, G.L. 9H-Carbazole-1-carboxamides as potent and selective JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 2809–2812. [Google Scholar] [CrossRef]
- Baburajeev, C.; Mohan, C.D.; Patil, G.S.; Rangappa, S.; Pandey, V.; Sebastian, A.; Fuchs, J.E.; Bender, A.; Lobie, P.E.; Rangappa, K.S. Nano-cuprous oxide catalyzed one-pot synthesis of a carbazole-based STAT3 inhibitor: A facile approach via intramolecular C–N bond formation reactions. RSC Adv. 2016, 6, 36775–36785. [Google Scholar] [CrossRef]
- Diaz, T.; Navarro, A.; Ferrer, G.; Gel, B.; Gaya, A.; Artells, R.; Bellosillo, B.; Garcia-Garcia, M.; Serrano, S.; Martínez, A. Lestaurtinib inhibition of the Jak/STAT signaling pathway in hodgkin lymphoma inhibits proliferation and induces apoptosis. PLoS ONE 2011, 6, e18856. [Google Scholar] [CrossRef]
- Shabbir, M.; Stuart, R. Lestaurtinib, a multitargeted tyrosinse kinase inhibitor: From bench to bedside. Expert Opin. Investig. Drugs 2010, 19, 427–436. [Google Scholar] [CrossRef]
- Joos, S.; Küpper, M.; Ohl, S.; Von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Möller, P.; Pfreundschuh, M.; Trümper, L. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Amarante-Mendes, G.P.; McGahon, A.J.; Nishioka, W.K.; Afar, D.E.; Witte, O.N.; Green, D.R. Bcl-2-independent Bcr–Abl-mediated resistance to apoptosis: Protection is correlated with up regulation of Bcl-x L. Oncogene 1998, 16, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.; Melzner, I.; Menz, C.; Wegener, S.; Bucur, A.; Dorsch, K.; Mattfeldt, T.; Barth, T.; Möller, P. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 2006, 25, 2679–2684. [Google Scholar] [CrossRef]
- Kube, D.; Holtick, U.; Vockerodt, M.; Ahmadi, T.; Haier, B.; Behrmann, I.; Heinrich, P.C.; Diehl, V.; Tesch, H. STAT3 is constitutively activated in Hodgkin cell lines. Blood 2001, 98, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.L.; Tibes, R.; Mesa, R.A. JAK2 inhibitors and their impact in myeloproliferative neoplasms. Hematology 2012, 17, s129–s132. [Google Scholar] [CrossRef]
- Santos, F.P.; Kantarjian, H.M.; Jain, N.; Manshouri, T.; Thomas, D.A.; Garcia-Manero, G.; Kennedy, D.; Estrov, Z.; Cortes, J.; Verstovsek, S. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010, 115, 1131–1136. [Google Scholar] [CrossRef]
- Issa, S.; Prandina, A.; Bedel, N.; Rongved, P.; Yous, S.; Le Brogne, M.; Bouaziz, Z. Carbazole scaffolds in cancer therapy: A review from 2012 to 2018. J. Enzym. Inhib Med. Chem 2019, 34, 1321–1346. [Google Scholar] [CrossRef]
- Tibes, R.; Bogenberger, J.M.; Benson, K.L.; Mesa, R.A. Current outlook on molecular pathogenesis and treatment of myeloproliferative neoplasm. Mol. Diagn. Ther. 2012, 16, 269–283. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name Compound | Structure Compound | References |
|---|---|---|---|
| 1 | Carbazole |  | Arbiser et al. [31] |
| 11 | 2-Hydroxycarbazole |  | Arbiser et al. [31] |
| 12 | Mahanine 3,11-dihydro-3,5-dimethyl-3-(4-methyl-3-pentenyl)-pyrano[3,2-a]carbazol-9-ol |  | Sarkar et al. [100] |
| 13a | Dimethyl-5-(5-(6-methoxy-1,4-dimethyl-9H-carbazol-9-yl) pentyloxy)isophthalate |  | Saturnino et al. [32] |
| 13b | Dimethyl-5-(6-(6-methoxy-1,4-dimethyl-9H-carbazol-9-yl) hexyloxy)isophthalate |  | Saturnino et al. [32] |
| 13c | Dimethyl-5-(7-(6-methoxy-1,4-dimethyl-9H-carbazol-9-yl) heptyloxy)isophthalate |  | Saturnino et al. [32] |
| 14a | Methyl-2-(5-(6-methoxy-1,4-dimethyl-9H-carbazol-9-yl) pentyloxy) benzoate |  | Saturnino et al. [32] |
| 14b | Methyl-2-(6-(6-methoxy-1,4-dimethyl-9H-carbazol-9-yl) hexyloxy)benzoate |  | Saturnino et al. [32] |
| 14c | Methyl-2-(7-(6-methoxy-1,4-dimethyl-9H-carbazol-9-yl) heptyloxy)benzoate |  | Saturnino et al. [32] |
| 15 | 7-Hydroxy-1-methyl-9H-carbazol-2-yl-5-(dimethylamino)-naphthalene-1-sulfonate |  | Hou et al. [107]; Kang et al. [111] |
| 16 | EC-70124 (5R,7S,9S)-7,8-Dihydroxy-9-methyl-6,7,8,9-tetrahydro-5H,14H-17-oxa-4b,9a,15-triaza-5,9-methanodibenzo[b,h]cyclonona[jkl]cyclopenta[e]-as-indacene-14,16(15H)-dione |  | Cuenca-López et al. [113] Estupiñan et al. [114] |
| 17 | 1,4-Dimethyl-9H-carbazole |  | Botta et al. [115] |
| 18 | 6-Hydroxy-1,4-dimethyl-9H-carbazole |  | Botta et al. [115] |
| 19 | 6-Methoxy-1,4-dimethyl-9H-carbazole |  | Botta et al. [115] |
| 20 | Ethyl-5,8-dimethyl-9H-carbazole-3-carboxylate |  | Botta et al. [115] |
| 21 | 6-Chloro-1,4-dimethyl-9H-carbazole |  | Botta et al. [115] |
| 22 | 5,8-Dimethyl-9H-carbazole-3-sulfonamide |  | Botta et al. [115] |
| 23 | 6-Methoxy-1,4-dimethyl-3-nitro-9H-carbazole |  | Botta et al. [115] |
| 24 | Ethyl-5,8-dimethyl-9H-carbazole-3-nitro-carboxylate |  | Botta et al. [115] |
| 25 | 6-Methoxy-1,4,9-trimethyl-carbazole |  | Botta et al. [115] |
| 26 | 6-Methoxy-1,4-dimethyl-9-ethyl-carbazole |  | Botta et al. [115] |
| 27 | 3-(3,4-Dicholophenyl)-6-(morpholine-4-carbonyl)-9H-carbazole-1-carboxamide |  | Zimmermann et al. [116] |
| 28 | (S)-7-(2-(dimethylamino)-pyrrolidine-1-carbonyl)-3-(3-methoxyphenyl)-9H-carbazole-1-carboxamide |  | Zimmermann et al. [116] |
| 29 | 3-(1-Methyl-1H-indazol-5-yl)-7-(4-methylpiperazin-1-yl)-9H-carbazole-1-carboxamide |  | Zimmermann et al. [116] |
| 30 | 3′-((3-Acetyl-6-chloro-9H-carbazol-9-yl)methyl)-[1,1′-biphenyl]-2-carbonitrile |  | Baburajeev et al. [117] |
| 31 | 3′-((3-Chloro-6-ethyl-9H-carbazol-9-yl)methyl)-[1,1′-biphenyl]-2-carbonitrile |  | Baburajeev et al. [117] |
| 32 | 3′-((3-Chloro-6-(1-hydroxyethyl)-9H-carbazol-9-yl)methyl)-[1,1′-biphenyl]-2-carbonitrile |  | Baburajeev et al. [117] |
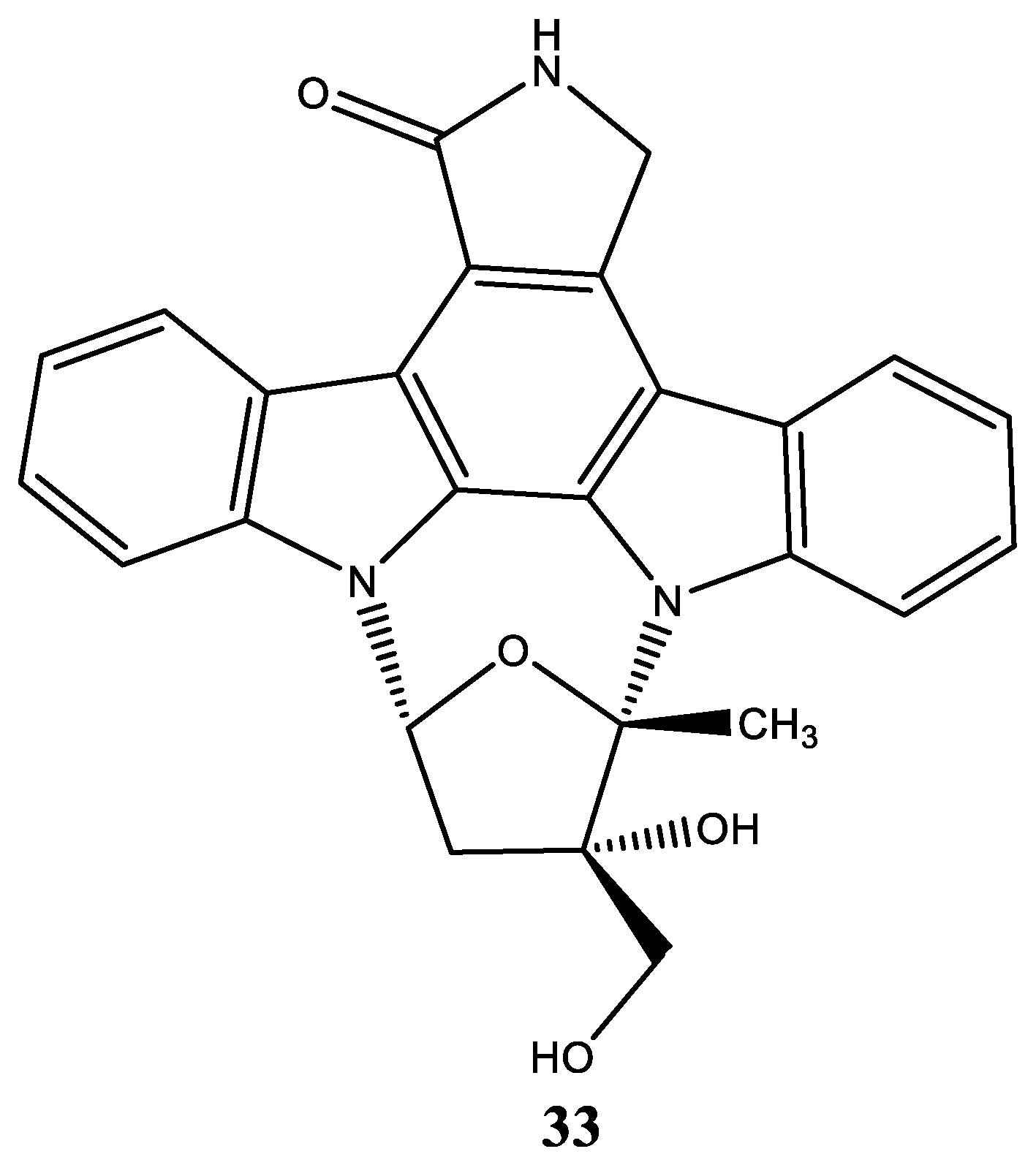
| 33 | CEP-701 Lestaurtinib (5S,6S,8R)-6-Hydroxy-6-(hydroxymethyl)-5-methyl-7,8,14,15-tetrahydro-5H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacen-13(6H)-one |  | Diaz et al. [118] Shabbir et al. [119] Geyer et al. [124] Santos et al. [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruso, A.; Barbarossa, A.; Carocci, A.; Salzano, G.; Sinicropi, M.S.; Saturnino, C. Carbazole Derivatives as STAT Inhibitors: An Overview. Appl. Sci. 2021, 11, 6192. https://doi.org/10.3390/app11136192
Caruso A, Barbarossa A, Carocci A, Salzano G, Sinicropi MS, Saturnino C. Carbazole Derivatives as STAT Inhibitors: An Overview. Applied Sciences. 2021; 11(13):6192. https://doi.org/10.3390/app11136192
Chicago/Turabian StyleCaruso, Anna, Alexia Barbarossa, Alessia Carocci, Giovanni Salzano, Maria Stefania Sinicropi, and Carmela Saturnino. 2021. "Carbazole Derivatives as STAT Inhibitors: An Overview" Applied Sciences 11, no. 13: 6192. https://doi.org/10.3390/app11136192
APA StyleCaruso, A., Barbarossa, A., Carocci, A., Salzano, G., Sinicropi, M. S., & Saturnino, C. (2021). Carbazole Derivatives as STAT Inhibitors: An Overview. Applied Sciences, 11(13), 6192. https://doi.org/10.3390/app11136192