
Genomic and Transcriptomic Profiling of Amino Acid Compositions in Common Carp Fillets

 , , and
, , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish Collection
2.2. AA Quantification in Fillets
2.3. Estimating the Heritabilities of the AA Contents
2.4. Genome-Wide Association Study
2.5. Transcriptomic Analysis to Identify DEGs Related to AA Contents
2.6. Core Genes of the THEAA Concentration and PPI Network Construction
2.7. qRT-PCR Validation of the Core Genes Related to THEAA Content
2.8. Genome Selection Analysis
3. Results
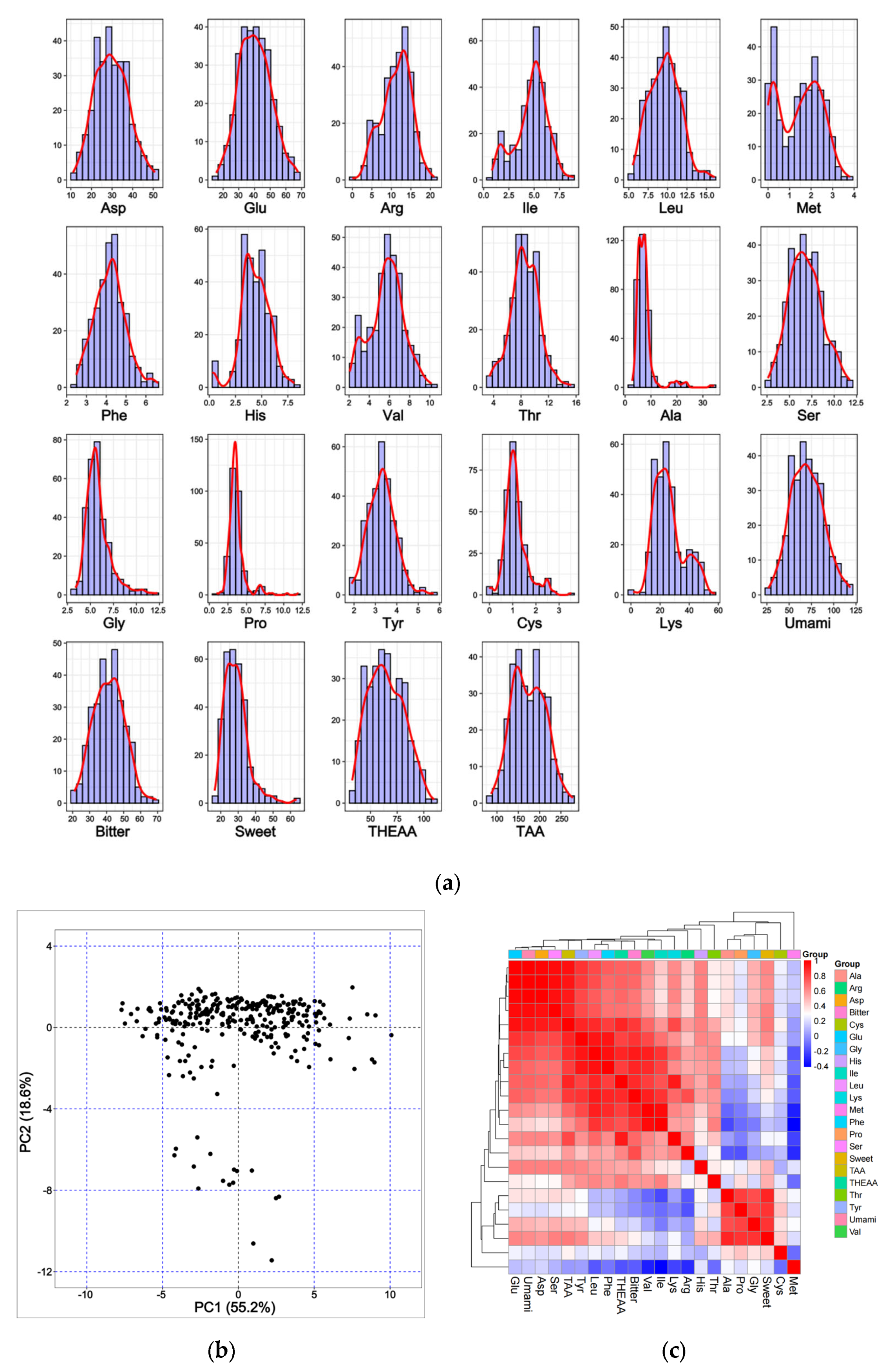
3.1. Heterogeneities in the Levels of Fillet AAs
3.2. High or Medium Heritabilities of AA Contents
3.3. Genetic Variants and Candidate Genes Associated with the AA Contents
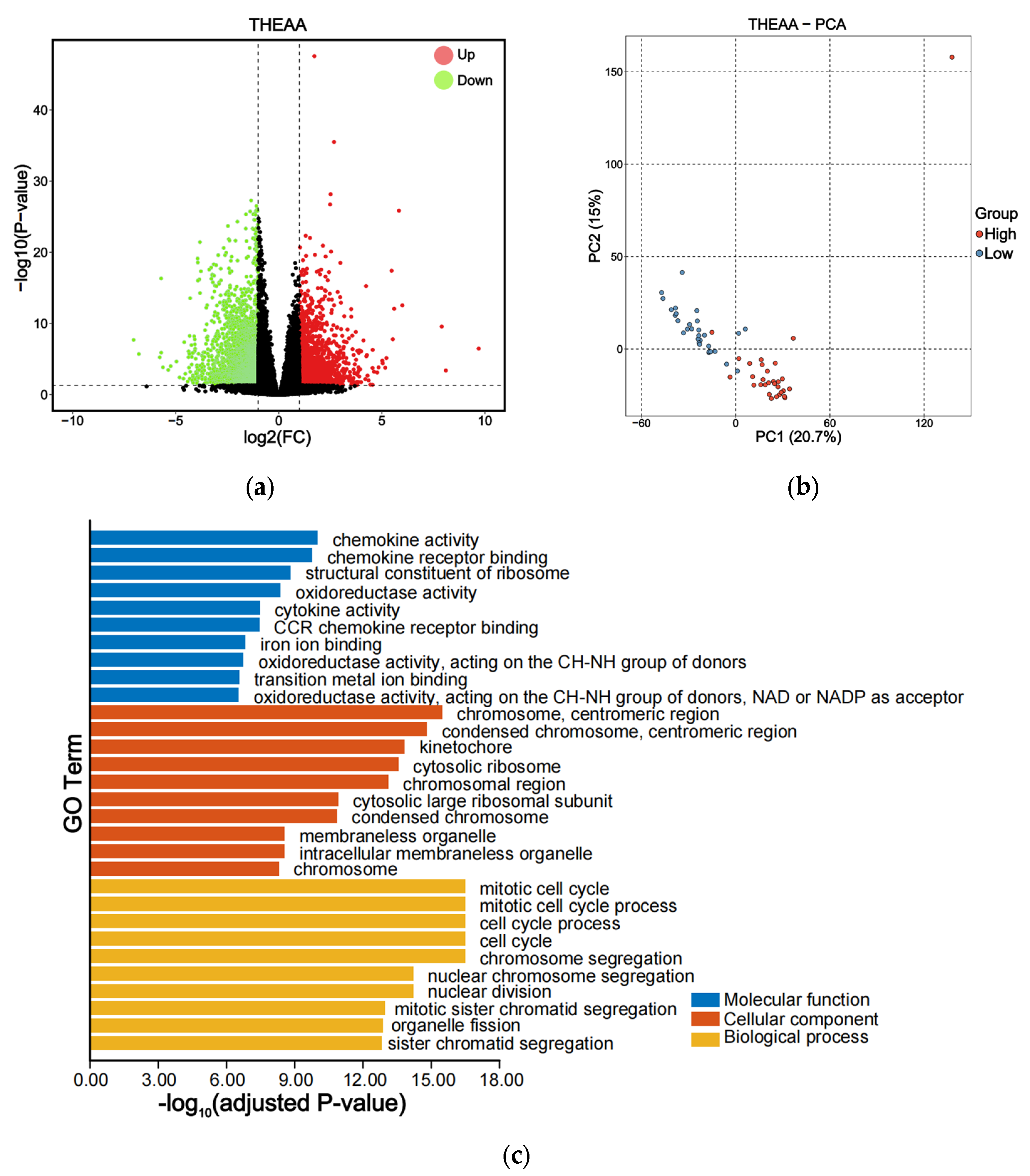
3.4. DEGs Related to the AA Concentrations
3.5. Core Genes to Different AA Concentrations by Integrating GWAS and RNA-Seq Analysis
3.6. qPCR Validated the Core Genes Related to THEAA Content
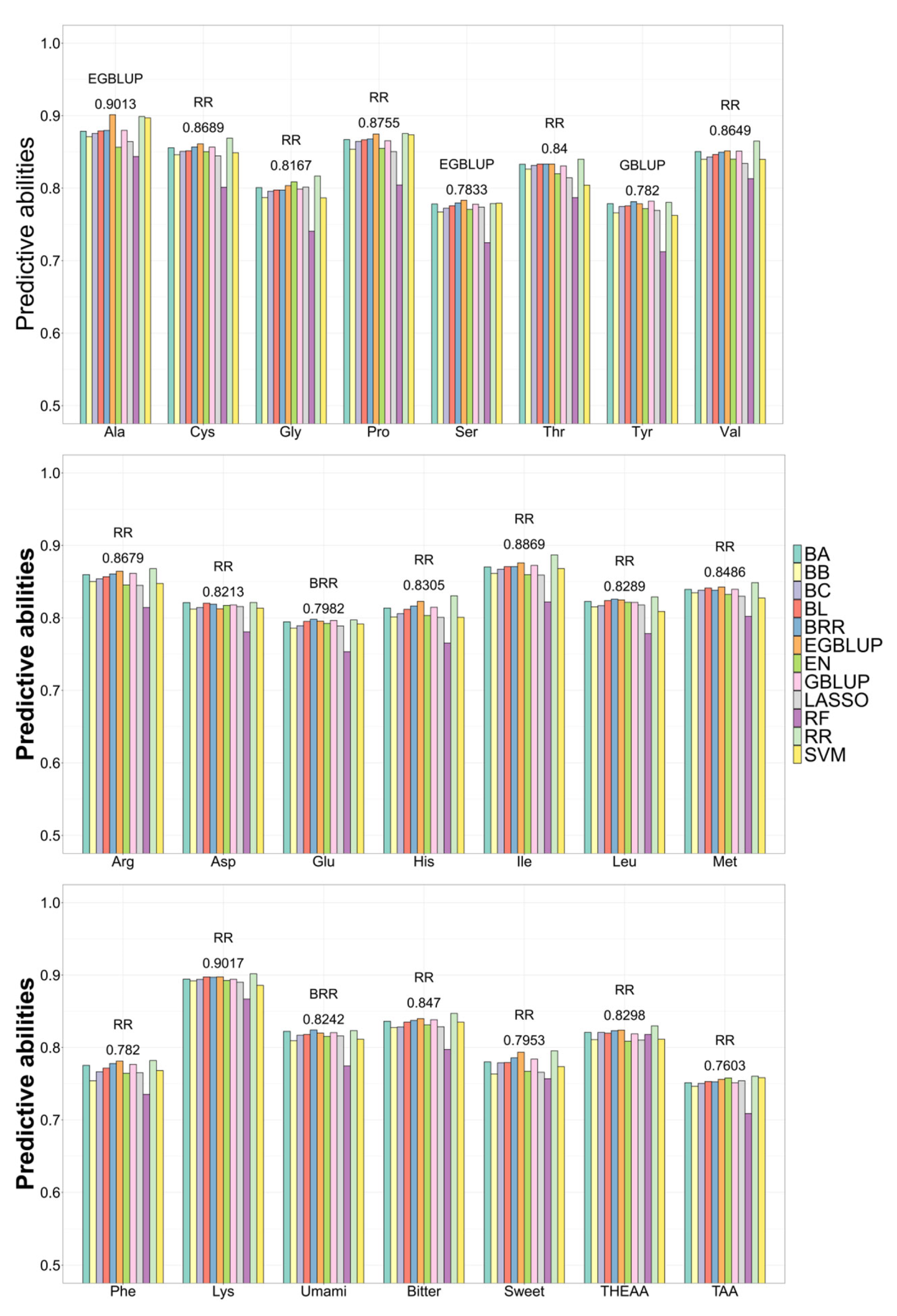
3.7. Application of GS to Predict AA Concentration
4. Discussion
4.1. Competition and Coordination in the Metabolism of Different AAs
4.2. Catalytic Activities of Oxidoreductases
4.3. Participation of Immune-Related Pathways to AA Metabolism
4.4. Application of GS and Gene-Editing Technique in Improving the Fillet AA Contents
4.5. Application of Other Omics Data in Identifying AA-Related Genes and Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAs | Amino acids |
| HEAAs | Human essential amino acids |
| THEAA | The concentration of total HEAA |
| TAA | Total amino acid |
References
- Mohanty, B.; Mahanty, A.; Ganguly, S.; Sankar, T.V.; Chakraborty, K.; Rangasamy, A.; Paul, B.; Sarma, D.; Mathew, S.; Asha, K.K.; et al. Amino Acid compositions of 27 food fishes and their importance in clinical nutrition. J. Amino Acids 2014, 2014, 269797. [Google Scholar] [CrossRef] [PubMed]
- Faber, T.A.; Bechtel, P.J.; Hernot, D.C.; Parsons, C.M.; Swanson, K.S.; Smiley, S.; Fahey, G.C., Jr. Protein digestibility evaluations of meat and fish substrates using laboratory, avian, and ileally cannulated dog assays. J. Anim. Sci. 2010, 88, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Aragão, C.; Gonçalves, A.T.; Costas, B.; Azeredo, R.; Xavier, M.J.; Engrola, S. Alternative Proteins for Fish Diets: Implications beyond Growth. Animals 2022, 12, 1211. [Google Scholar] [CrossRef] [PubMed]
- Glencross, B.; Baily, J.; Berntssen, M.; Hardy, R.; Mackenzie, S.; Tocher, D. Risk assessment of the use of alternative animal and plant raw material resources in aquaculture feeds. Rev. Aquac. 2019, 12, 703–758. [Google Scholar] [CrossRef]
- Kim, W.J.; Kang, B.H.; Kang, S.; Shin, S.; Chowdhury, S.; Jeong, S.C.; Choi, M.S.; Park, S.K.; Moon, J.K.; Ryu, J.; et al. A Genome-Wide Association Study of Protein, Oil, and Amino Acid Content in Wild Soybean (Glycine soja). Plants 2023, 12, 1665. [Google Scholar] [CrossRef]
- Qin, J.; Shi, A.; Song, Q.; Li, S.; Wang, F.; Cao, Y.; Ravelombola, W.; Song, Q.; Yang, C.; Zhang, M. Genome Wide Association Study and Genomic Selection of Amino Acid Concentrations in Soybean Seeds. Front. Plant Sci. 2019, 10, 1445. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, H.; Chen, J.; Shi, T.; Zhang, C.; Sun, D.; He, Z.; Hao, Y.; Chen, W. Genome-Wide Association Studies of Free Amino Acid Levels by Six Multi-Locus Models in Bread Wheat. Front. Plant Sci. 2018, 9, 1196. [Google Scholar] [CrossRef]
- Deng, M.; Li, D.; Luo, J.; Xiao, Y.; Liu, H.; Pan, Q.; Zhang, X.; Jin, M.; Zhao, M.; Yan, J. The genetic architecture of amino acids dissection by association and linkage analysis in maize. Plant Biotechnol. J. 2017, 15, 1250–1263. [Google Scholar] [CrossRef]
- Wang, R.; Lu, Y.; Qi, J.; Xi, Y.; Shen, Z.; Twumasi, G.; Bai, L.; Hu, J.; Wang, J.; Li, L.; et al. Genome-wide association analysis explores the genetic loci of amino acid content in duck’s breast muscle. BMC Genom. 2024, 25, 486. [Google Scholar] [CrossRef]
- Ali, A.; Al-Tobasei, R.; Lourenco, D.; Leeds, T.; Kenney, B.; Salem, M. Genome-Wide Association Study Identifies Genomic Loci Affecting Filet Firmness and Protein Content in Rainbow Trout. Front. Genet. 2019, 10, 386. [Google Scholar] [CrossRef]
- Ling, Z.N.; Jiang, Y.F.; Ru, J.N.; Lu, J.H.; Ding, B.; Wu, J. Amino acid metabolism in health and disease. Signal Transduct. Target. Ther. 2023, 8, 345. [Google Scholar] [CrossRef] [PubMed]
- Freidman, N.; Chen, I.; Wu, Q.; Briot, C.; Holst, J.; Font, J.; Vandenberg, R.; Ryan, R. Amino Acid Transporters and Exchangers from the SLC1A Family: Structure, Mechanism and Roles in Physiology and Cancer. Neurochem. Res. 2020, 45, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Moitra, K.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Hum. Mutat. 2022, 43, 1162–1182. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Peng, H.F.; Zhang, X.; Zhang, H.S. Comprehensive review of amino acid transporters as therapeutic targets. Int. J. Biol. Macromol. 2024, 260, 129646. [Google Scholar] [CrossRef]
- Guedes, R.L.; Prosdocimi, F.; Fernandes, G.R.; Moura, L.K.; Ribeiro, H.A.; Ortega, J.M. Amino acids biosynthesis and nitrogen assimilation pathways: A great genomic deletion during eukaryotes evolution. BMC Genom. 2011, 12 (Suppl. S4), S2. [Google Scholar] [CrossRef]
- Torres, N.; Tobón-Cornejo, S.; Velazquez-Villegas, L.A.; Noriega, L.G.; Alemán-Escondrillas, G.; Tovar, A.R. Amino Acid Catabolism: An Overlooked Area of Metabolism. Nutrients 2023, 15, 3378. [Google Scholar] [CrossRef]
- Nakajima, T.; Hudson, M.J.; Uchiyama, J.; Makibayashi, K.; Zhang, J. Common carp aquaculture in Neolithic China dates back 8000 years. Nat. Ecol. Evol. 2019, 3, 1415–1418. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, Y.; Xu, P.; Zhao, Z.; Feng, J.; Wu, B.; Jiang, Y.; Dong, C.; Xu, J. Multi-Omics Data Reveal Amino Acids Related Genes in the Common Carp Cyprinus carpio. Fishes 2022, 7, 225. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Cao, Y.-M.; Hou, M.-X.; Zhao, R.; Chen, Y.-J.; Yu, S.-T.; Wang, K.-K.; Zhang, Q.; Li, S.-J.; et al. Genome-Wide Association Analysis Identifies Genetic Variants Associated with Muscle Fatty Acids and Amino Acids in Grass Carp (Ctenopharyngodon idella). Agric. Commun. 2024, 2, 100043. [Google Scholar] [CrossRef]
- Schmidt, C.V.; Olsen, K.; Mouritsen, O.G. Umami synergy as the scientific principle behind taste-pairing champagne and oysters. Sci. Rep. 2020, 10, 20077. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tong, H. An overview of bitter compounds in foodstuffs: Classifications, evaluation methods for sensory contribution, separation and identification techniques, and mechanism of bitter taste transduction. Compr. Rev. Food Sci. Food Saf. 2023, 22, 187–232. [Google Scholar] [CrossRef] [PubMed]
- Reis, G.C.L.; Guidi, L.R.; Fernandes, C.; Godoy, H.T.; Gloria, M.B.A. UPLC-UV Method for the Quantification of Free Amino Acids, Bioactive Amines, and Ammonia in Fresh, Cooked, and Canned Mushrooms. Food Anal. Methods 2020, 13, 1613–1626. [Google Scholar] [CrossRef]
- Garip, B.; Khokhar, J.Y.; Kayir, H. Plasma essential amino acid levels in first episode psychosis at baseline and after antipsychotic treatment. Schizophrenia 2024, 10, 103. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2 Elegant Graphics for Data Analysis. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Irnawati, I.; Riswanto, F.; Riyanto, S.; Martono, S.; Rohman, A. The use of software packages of R factoextra and FactoMineR and their application in principal component analysis for authentication of oils. Indones. J. Chemom. Pharm. Anal. 2020, 1, 1–10. [Google Scholar] [CrossRef]
- Li, J.T.; Wang, Q.; Huang Yang, M.D.; Li, Q.S.; Cui, M.S.; Dong, Z.J.; Wang, H.W.; Yu, J.H.; Zhao, Y.J.; Yang, C.R.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Yin, D.; Fu, Y.; Yuan, X.; Li, X.; Liu, X.; Zhao, S. HIBLUP: An integration of statistical models on the BLUP framework for efficient genetic evaluation using big genomic data. Nucleic Acids Res. 2023, 51, 3501–3512. [Google Scholar] [CrossRef]
- Adhikari, B.; Joshi, B.; Shrestha, J.; Bhatta, N. Genetic variability, heritability, genetic advance and correlation among yield and yield components of rice. J. Agric. Nat. Resour. 2018, 1, 149–160. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Shim, H.; Chasman, D.I.; Smith, J.D.; Mora, S.; Ridker, P.M.; Nickerson, D.A.; Krauss, R.M.; Stephens, M. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS ONE 2015, 10, e0120758. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nature Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Charmet, G.; Tran, L.G.; Auzanneau, J.; Rincent, R.; Bouchet, S. BWGS: A R package for genomic selection and its application to a wheat breeding programme. PLoS ONE 2020, 15, e0222733. [Google Scholar] [CrossRef] [PubMed]
- Højrup, P. Analysis of Peptides and Conjugates by Amino Acid Analysis. Methods Mol. Biol. 2015, 1348, 65–76. [Google Scholar] [CrossRef]
- Björkeroth, J.; Campbell, K.; Malina, C.; Yu, R.; Di Bartolomeo, F.; Nielsen, J. Proteome reallocation from amino acid biosynthesis to ribosomes enables yeast to grow faster in rich media. Proc. Natl. Acad. Sci. USA 2020, 117, 21804–21812. [Google Scholar] [CrossRef]
- Gómez-Tamayo, J.C.; Cordomí, A.; Olivella, M.; Mayol, E.; Fourmy, D.; Pardo, L. Analysis of the interactions of sulfur-containing amino acids in membrane proteins. Protein Sci. A Publ. Protein Soc. 2016, 25, 1517–1524. [Google Scholar] [CrossRef]
- Castellano, R.; Perruchot, M.H.; Tesseraud, S.; Métayer-Coustard, S.; Baeza, E.; Mercier, Y.; Gondret, F. Methionine and cysteine deficiencies altered proliferation rate and time-course differentiation of porcine preadipose cells. Amino Acids 2017, 49, 355–366. [Google Scholar] [CrossRef]
- Kanai, Y.; Fukasawa, Y.; Cha, S.H.; Segawa, H.; Chairoungdua, A.; Kim, D.K.; Matsuo, H.; Kim, J.Y.; Miyamoto, K.; Takeda, E.; et al. Transport properties of a system y+L neutral and basic amino acid transporter. Insights into the mechanisms of substrate recognition. J. Biol. Chem. 2000, 275, 20787–20793. [Google Scholar] [CrossRef]
- Das, P.; Sen, P. Relevance of Oxidoreductases in Cellular Metabolism and Defence. In Reactive Oxygen Species—Advances and Developments; Ahmad, R., Ed.; IntechOpen: Rijeka, Croatia, 2024. [Google Scholar]
- Sellés Vidal, L.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef]
- Brunhuber, N.M.; Thoden, J.B.; Blanchard, J.S.; Vanhooke, J.L. Rhodococcus L-phenylalanine dehydrogenase: Kinetics, mechanism, and structural basis for catalytic specificity. Biochemistry 2000, 39, 9174–9187. [Google Scholar] [CrossRef]
- Stillman, T.J.; Baker, P.J.; Britton, K.L.; Rice, D.W. Conformational flexibility in glutamate dehydrogenase. Role of water in substrate recognition and catalysis. J. Mol. Biol. 1993, 234, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.L.; Facey, C.O.B.; Boman, B.M. The Significance of Aldehyde Dehydrogenase 1 in Cancers. Int. J. Mol. Sci. 2024, 26, 251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bie, J.; Song, C.; Liu, M.; Luo, J. PYCR, a key enzyme in proline metabolism, functions in tumorigenesis. Amino Acids 2021, 53, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Hitzel, J.; Lee, E.; Zhang, Y.; Bibli, S.I.; Li, X.; Zukunft, S.; Pflüger, B.; Hu, J.; Schürmann, C.; Vasconez, A.E.; et al. Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells. Nat. Commun. 2018, 9, 2292. [Google Scholar] [CrossRef]
- Watcharanurak, K.; Zang, L.; Nishikawa, M.; Yoshinaga, K.; Yamamoto, Y.; Takahashi, Y.; Ando, M.; Saito, K.; Watanabe, Y.; Takakura, Y. Effects of upregulated indoleamine 2, 3-dioxygenase 1 by interferon γ gene transfer on interferon γ-mediated antitumor activity. Gene Ther. 2014, 21, 794–801. [Google Scholar] [CrossRef]
- Correale, J. Immunosuppressive Amino-Acid Catabolizing Enzymes in Multiple Sclerosis. Front. Immunol. 2020, 11, 600428. [Google Scholar] [CrossRef]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef]
- Kelly, B.; Pearce, E.L. Amino Assets: How Amino Acids Support Immunity. Cell Metab. 2020, 32, 154–175. [Google Scholar] [CrossRef]
- Howie, D.; Waldmann, H.; Cobbold, S. Nutrient Sensing via mTOR in T Cells Maintains a Tolerogenic Microenvironment. Front. Immunol. 2014, 5, 409. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, Q.; Xu, Y.; Feng, D.; Fei, J.; Cheng, Q.; Xu, L. Gamma-aminobutyric acid transporter 1 negatively regulates T cell activation and survival through protein kinase C-dependent signaling pathways. J. Immunol. 2009, 183, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Zhang, N.-N.; Fan, W.-J.; Cui, Y.-Y.; Limbu, S.; Qiao, F.; Zhao, Y.-L.; Chen, L.-Q.; Du, Z.-Y.; Li, D. Soybean and cottonseed meals are good candidates for fishmeal replacement in the diet of juvenile Macrobrachium nipponense. Aquac. Int. 2018, 26, 309–324. [Google Scholar] [CrossRef]
- Stock, K.F.; Reents, R. Genomic selection: Status in different species and challenges for breeding. Reprod. Domest. Anim. Zuchthyg. 2013, 48 (Suppl. S1), 2–10. [Google Scholar] [CrossRef]
- e Sousa, M.B.; Galli, G.; Lyra, D.H.; Granato, Í.S.C.; Matias, F.I.; Alves, F.C.; Fritsche-Neto, R. Increasing accuracy and reducing costs of genomic prediction by marker selection. Euphytica 2019, 215, 18. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, Y.; Zhang, J.-F.; Bi, W. Efficient and accurate framework for genome-wide gene-environment interaction analysis in large-scale biobanks. Nat. Commun. 2025, 16, 3064. [Google Scholar] [CrossRef]
- Javaid, D.; Ganie, S.Y.; Hajam, Y.A.; Reshi, M.S. CRISPR/Cas9 system: A reliable and facile genome editing tool in modern biology. Mol. Biol. Rep. 2022, 49, 12133–12150. [Google Scholar] [CrossRef]
- Shi, T.; Zhu, A.; Jia, J.; Hu, X.; Chen, J.; Liu, W.; Ren, X.; Sun, D.; Fernie, A.R.; Cui, F.; et al. Metabolomics analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. Cell Mol. Biol. 2020, 103, 279–292. [Google Scholar] [CrossRef]
- Lamri, M.; Della Malva, A.; Djenane, D.; López-Pedrouso, M.; Franco, D.; Albenzio, M.; Lorenzo, J.M.; Gagaoua, M. Towards the discovery of goat meat quality biomarkers using label-free proteomics. J. Proteom. 2023, 278, 104868. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Liu, C.; Birchler, J.A.; Han, F. Prospects and challenges of epigenomics in crop improvement. Genes Genom. 2022, 44, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Khunsriraksakul, C.; Markus, H.; Chen, D.; Zhang, F.; Chen, F.; Zhan, X.; Carrel, L.; Liu, D.J.; Jiang, B. Integrating single cell expression quantitative trait loci summary statistics to understand complex trait risk genes. Nat. Commun. 2024, 15, 4260. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, H.S. Amino acid metabolism, redox balance and epigenetic regulation in cancer. FEBS J. 2024, 291, 412–429. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean | SD | Max | Min | CV | |
|---|---|---|---|---|---|
| Asp | 29.73 | 7.98 | 50.82 | 11.01 | 27% |
| Glu | 40.29 | 10.05 | 67.81 | 15.57 | 25% |
| Arg | 11.26 | 3.76 | 20.99 | 0.00 | 33% |
| Ile | 4.73 | 1.60 | 8.93 | 0.82 | 34% |
| Leu | 9.64 | 1.97 | 16.06 | 5.51 | 20% |
| Met | 1.47 | 0.97 | 3.81 | 0.00 | 66% |
| Phe | 4.19 | 0.78 | 6.62 | 2.49 | 19% |
| His | 4.38 | 1.36 | 8.12 | 0.29 | 31% |
| Val | 5.74 | 1.66 | 10.18 | 2.06 | 29% |
| Thr | 8.54 | 2.04 | 15.31 | 3.40 | 24% |
| Ala | 7.59 | 3.99 | 34.38 | 2.75 | 53% |
| Ser | 6.81 | 1.83 | 11.75 | 2.60 | 27% |
| Gly | 5.86 | 1.39 | 12.29 | 3.38 | 24% |
| Pro | 3.64 | 1.18 | 11.98 | 0.96 | 32% |
| Tyr | 3.30 | 0.63 | 5.72 | 1.89 | 19% |
| Cys | 1.14 | 0.50 | 3.53 | 0.00 | 44% |
| Lys | 26.07 | 10.28 | 57.73 | 1.52 | 39% |
| Umami AA | 70.02 | 17.96 | 118.63 | 26.58 | 26% |
| Bitter AA | 41.41 | 8.98 | 69.25 | 21.06 | 22% |
| Sweet AA | 32.44 | 7.96 | 75.48 | 18.66 | 25% |
| TAA | 174.38 | 38.50 | 278.36 | 86.08 | 22% |
| THEAA | 64.76 | 16.45 | 109.69 | 32.82 | 25% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Wang, K.; Wang, Q.; Cao, Y.; Zhao, R.; Zhang, Y.; Li, J. Genomic and Transcriptomic Profiling of Amino Acid Compositions in Common Carp Fillets. Animals 2025, 15, 1335. https://doi.org/10.3390/ani15091335
Chen Y, Wang K, Wang Q, Cao Y, Zhao R, Zhang Y, Li J. Genomic and Transcriptomic Profiling of Amino Acid Compositions in Common Carp Fillets. Animals. 2025; 15(9):1335. https://doi.org/10.3390/ani15091335
Chicago/Turabian StyleChen, Yingjie, Kaikuo Wang, Qi Wang, Yiming Cao, Ran Zhao, Yan Zhang, and Jiongtang Li. 2025. "Genomic and Transcriptomic Profiling of Amino Acid Compositions in Common Carp Fillets" Animals 15, no. 9: 1335. https://doi.org/10.3390/ani15091335
APA StyleChen, Y., Wang, K., Wang, Q., Cao, Y., Zhao, R., Zhang, Y., & Li, J. (2025). Genomic and Transcriptomic Profiling of Amino Acid Compositions in Common Carp Fillets. Animals, 15(9), 1335. https://doi.org/10.3390/ani15091335