
Dynamic Changes in the Gut Microbiota During Peripartum in Jennies

 ,
,
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Animal Welfare Statement
2.2. Animals
2.3. Sampling
2.4. Samples’ Processing and 16S rRNA High-Throughput Sequencing
2.5. Bioinformatics and Statistical Analysis
3. Results
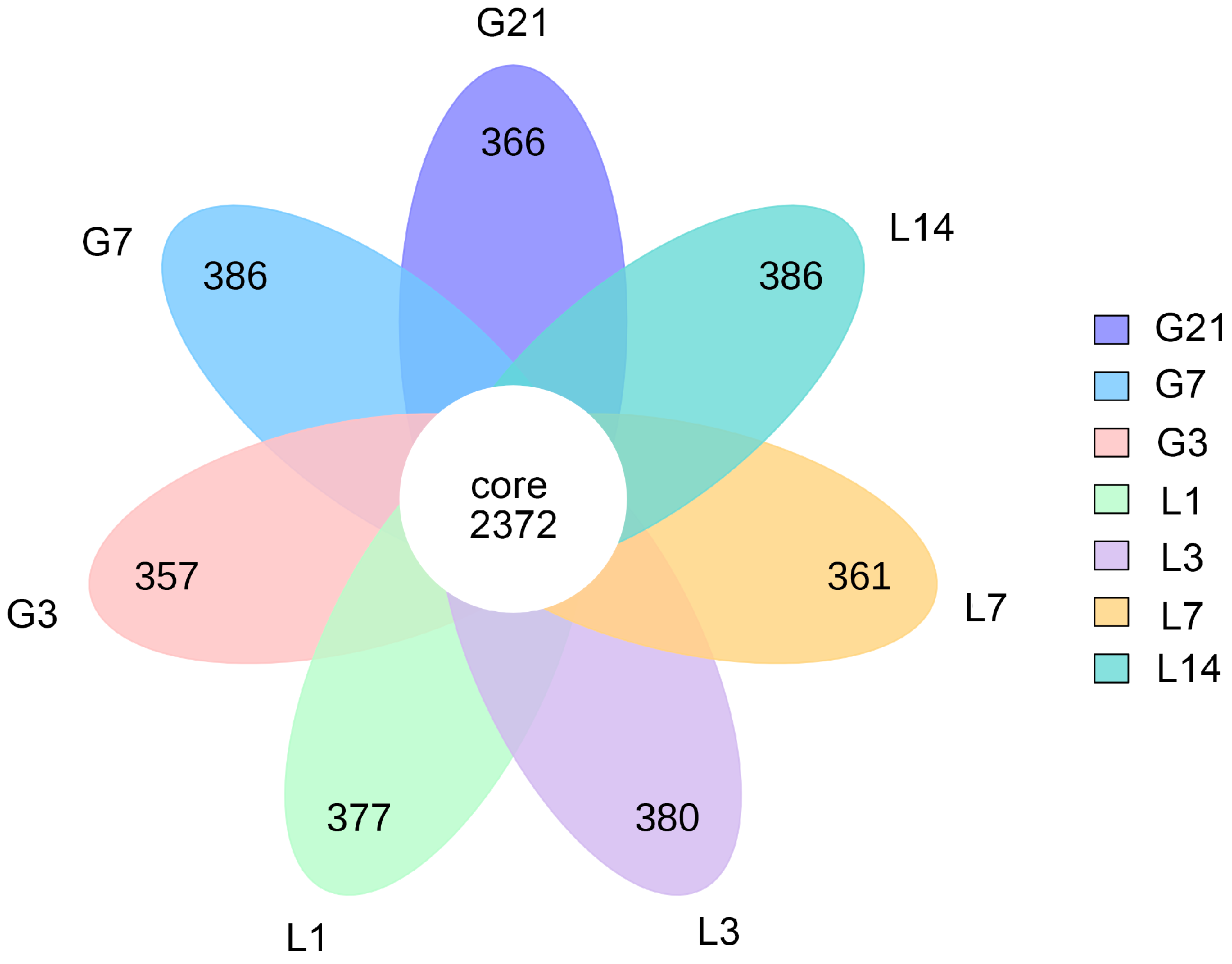
3.1. Sequencing Information
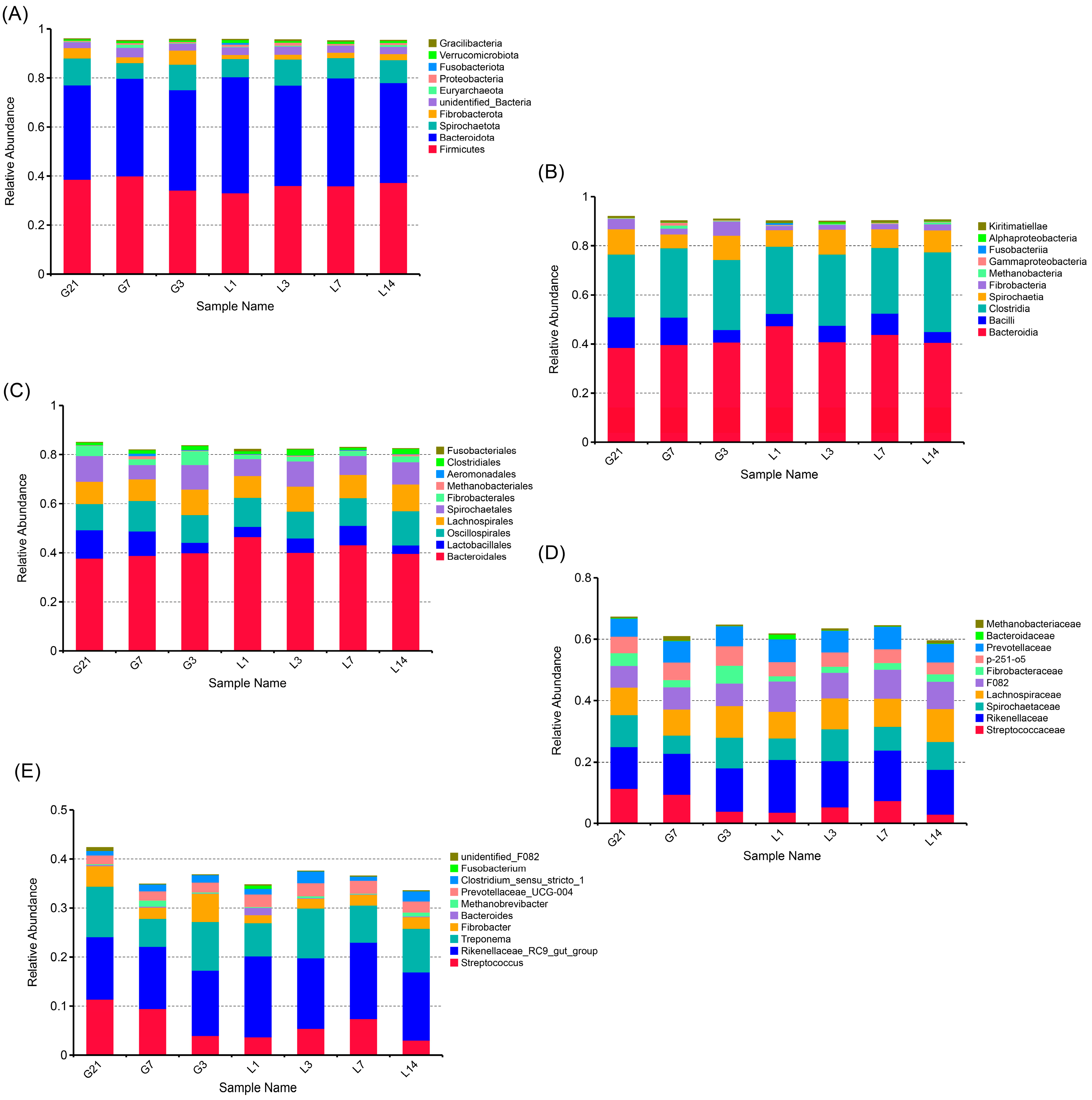
3.2. Taxonomic Composition of the Bacterial Communities
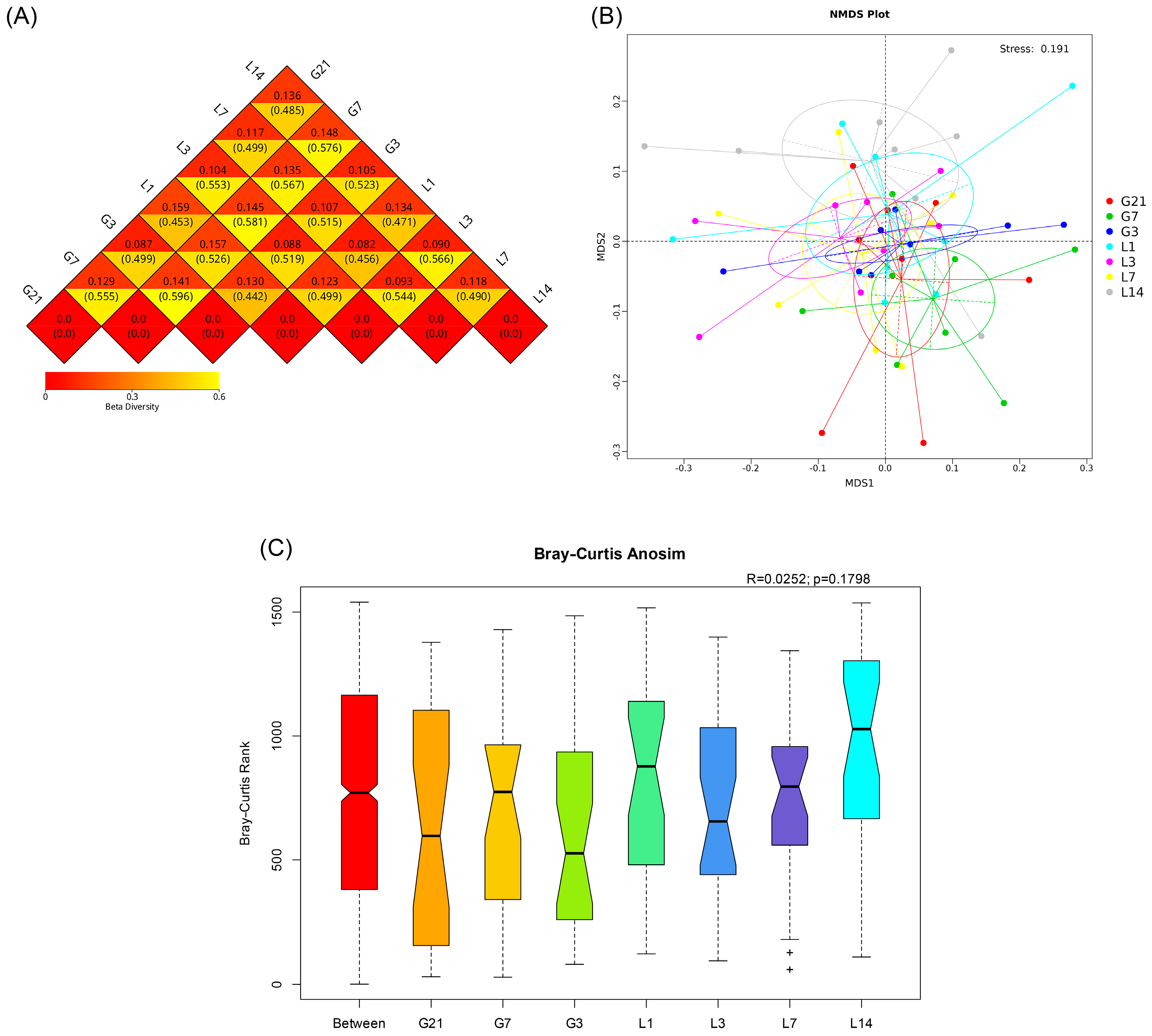
3.3. Alpha Diversity
3.4. Beta Diversity
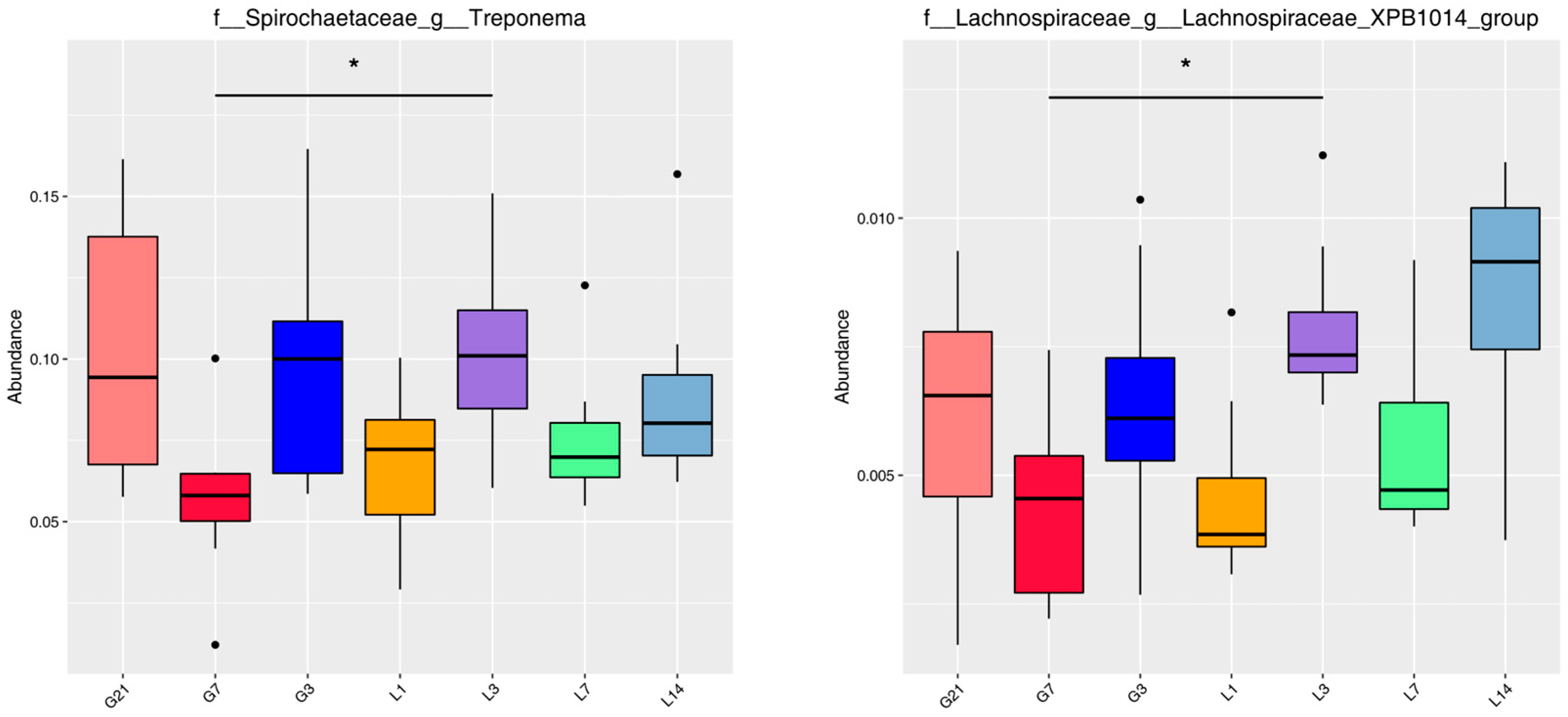
3.5. MetaStat Analysis
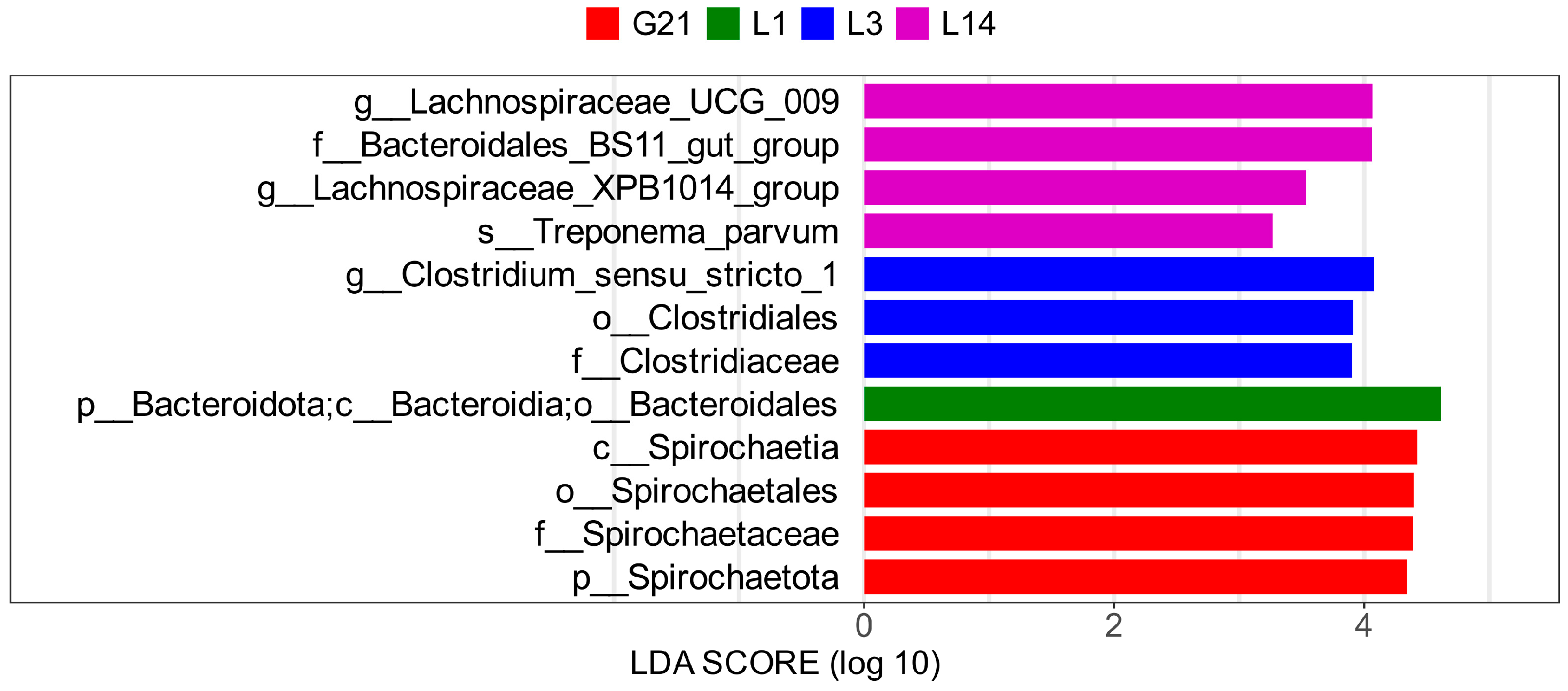
3.6. LDA Effect Size (LEfSe) Analysis
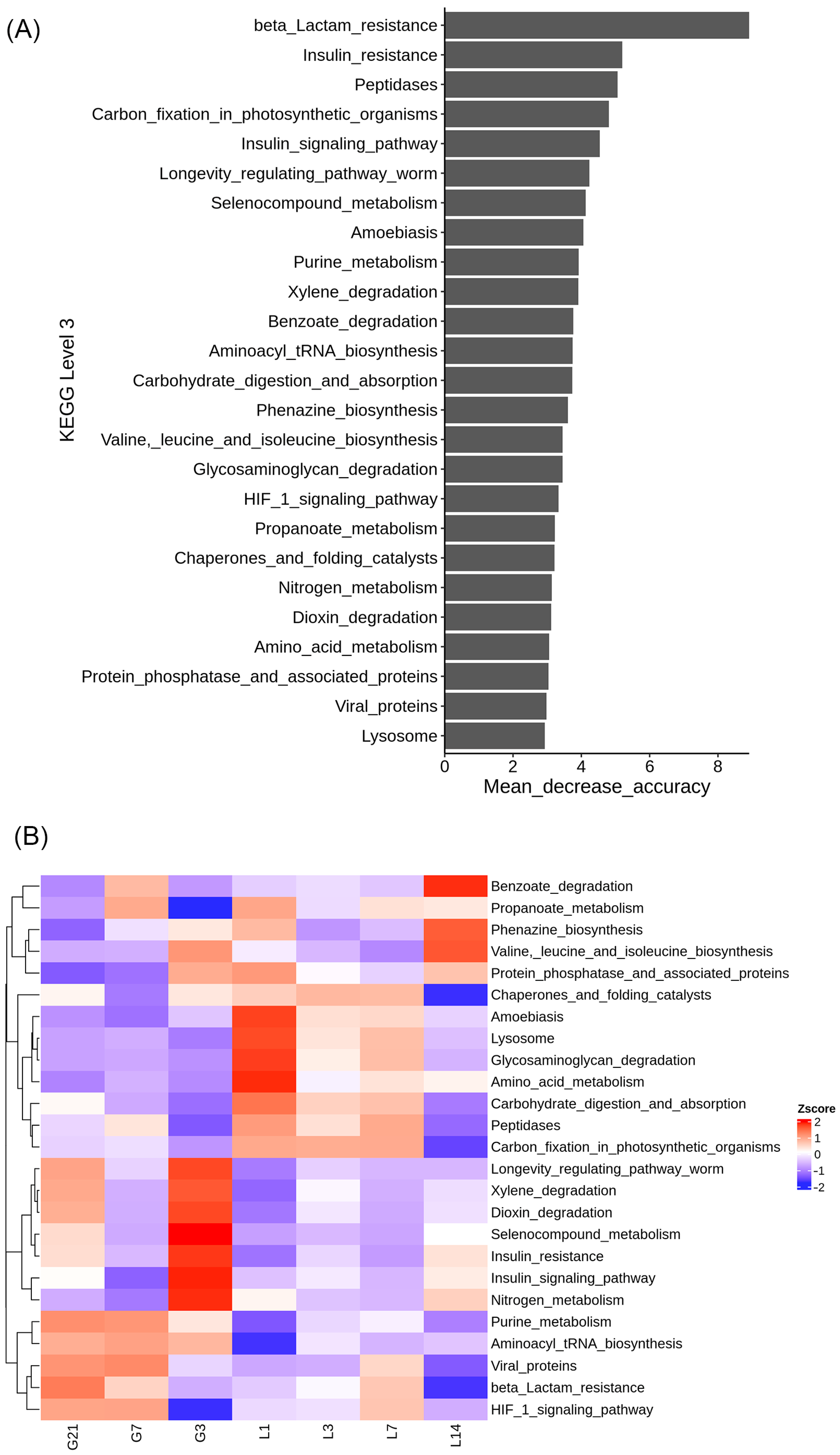
3.7. Functional Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, R.; Zhang, W.; Shen, W.; Zhang, G.; Xie, T.; Li, L.; Jinmei, J.; Liu, Y.; Kong, F.; Guo, B.; et al. Analysis of Gut Microbiota in Chinese Donkey in Different Regions Using Metagenomic Sequencing. BMC Genom. 2023, 24, 524. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; He, M.; Zhong, C.; Li, Y.; Tang, S.; Kang, X.; Wu, Z. Responses of Gut Microbiota in Crocodile Lizards (Shinisaurus crocodilurus) to Changes in Temperature. Front. Microbiol. 2023, 14, 1263917. [Google Scholar] [CrossRef]
- Luthold, R.V.; Fernandes, G.R.; Franco-de-Moraes, A.C.; Folchetti, L.G.D.; Ferreira, S.R.G. Gut Microbiota Interactions with the Immunomodulatory Role of Vitamin D in Normal Individuals. Metabolism 2017, 69, 76–86. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, H.; Jafari, H.; Liu, B.; Wang, Z.; Su, J.; Wang, F.; Yang, G.; Sun, M.; Cheng, J.; et al. Metabolic Changes before and after Weaning in Dezhou Donkey Foals in Relation to Gut Microbiota. Front. Microbiol. 2023, 14, 1306039. [Google Scholar] [CrossRef]
- Bao, W.; Yu, J.; He, Y.; Liu, M.; Yang, X. The Diversity Analysis and Gene Function Prediction of Intestinal Bacteria in Three Equine Species. Front. Microbiol. 2022, 13, 973828. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.E.; Schennink, A.; Burden, F.; Long, S.; van Doorn, D.A.; Pellikaan, W.F.; Dijkstra, J.; Saccenti, E.; Smidt, H. Domesticated Equine Species and Their Derived Hybrids Differ in Their Fecal Microbiota. Anim. Microbiome 2020, 2, 8. [Google Scholar] [CrossRef]
- Gao, H.; Jiang, F.; Zhang, J.; Chi, X.; Song, P.; Li, B.; Cai, Z.; Zhang, T. Effects of Ex Situ Conservation on Diversity and Function of the Gut Microbiota of the Tibetan Wild Ass (Equus kiang). Integr. Zool. 2023, 18, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Khan, M.Z.; Chen, Y.; Liang, H.; Kou, X.; Wang, X.; Ren, W.; Wang, C.; Zhang, Z. Yeast Polysaccharide Supplementation: Impact on Lactation, Growth, Immunity, and Gut Microbiota in Dezhou Donkeys. Front. Microbiol. 2023, 14, 1289371. [Google Scholar] [CrossRef]
- Bonelli, F.; Rota, A.; Corazza, M.; Serio, D.; Sgorbini, M. Hematological and Biochemical Findings in Pregnant, Postfoaling, and Lactating Jennies. Theriogenology 2016, 85, 1233–1238. [Google Scholar] [CrossRef]
- Amato, K.R.; Pradhan, P.; Mallott, E.K.; Shirola, W.; Lu, A. Host-Gut Microbiota Interactions during Pregnancy. Evol. Med. Public Health 2024, 12, 7–23. [Google Scholar] [CrossRef]
- Guo, Y.; Yin, G.; Hui, F.; Guo, X.; Shi, B.; Zhao, Y.; Yan, S. Effects of Dietary Energy Level on Antioxidant Capability, Immune Function and Rectal Microbiota in Late Gestation Donkeys. Front. Microbiol. 2024, 15, 1308171. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Li, Z.; Han, Y.; Deng, L. Comparative Analysis of Serum Mineral and Biochemical Parameter Profiles Between Late Pregnant and Early Lactating Jennies. J. Equine Vet. Sci. 2021, 99, 103401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, B.; Wang, Y.; Zhan, Y.; Zhu, M.; Wang, C. Dynamic Alterations in the Donkey Fecal Bacteria Community and Metabolome Characteristics during Gestation. Front. Microbiol. 2022, 13, 927561. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Donkeys and other equids. In Nutrient Requirements of Horses, 6th ed.; The National Academies Press: Washington, DC, USA, 2007. [Google Scholar]
- The Donkey Sanctuary. The clinical examination. In The Clinical Companion of the Donkey; Evans, L., Crane, M., Eds.; Matador Publishing Inc.: Leicestershire, UK, 2018; p. 256. [Google Scholar]
- Veronesi, M.C.; Gloria, A.; Panzani, S.; Sfirro, M.P.; Carluccio, A.; Contri, A. Blood Analysis in Newborn Donkeys: Hematology, Biochemistry, and Blood Gases Analysis. Theriogenology 2014, 82, 294–303. [Google Scholar] [CrossRef]
- Sharma, R.; John, S.J.; Damgaard, M.; McAllister, T.A. Extraction of PCR-Quality Plant and Microbial DNA from Total Rumen Contents. Biotechniques 2003, 34, 92–94, 96–97. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-High-Throughput Microbial Community Analysis on the Illumina HiSeq and MiSeq Platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Pyles, M.B.; Fowler, A.L.; Bill, V.T.; Harlow, B.E.; Crum, A.D.; Hayes, S.H.; Flythe, M.D.; Lawrence, L.M. Effect of Maternal Diet on Select Fecal Bacteria of Foals. Transl. Anim. Sci. 2019, 3, 204–211. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Q.; Liu, G.; Zhang, Z.; Zhan, Y.; Zhu, M.; Wang, C. Metabolic Alternations During Gestation in Dezhou Donkeys and the Link to the Gut Microbiota. Front. Microbiol. 2022, 13, 801976. [Google Scholar] [CrossRef]
- Stewart, H.L.; Pitta, D.; Indugu, N.; Vecchiarelli, B.; Engiles, J.B.; Southwood, L.L. Characterization of the Fecal Microbiota of Healthy Horses. Am. J. Vet. Res. 2018, 79, 811–819. [Google Scholar] [CrossRef]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stämpfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the Fecal Microbiota of Healthy Horses and Horses with Colitis by High Throughput Sequencing of the V3-V5 Region of the 16S rRNA Gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of Bacterial DNA Extraction Method from Fecal Material Influences Community Structure as Evaluated by Metagenomic Analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Azhar, E.I.; Abbas, A.T.; Kumosani, T.; Barbour, E.K.; Raoult, D.; Yasir, M. Metagenomic Analysis of Antibiotic-Induced Changes in Gut Microbiota in a Pregnant Rat Model. Front. Pharmacol. 2016, 7, 104. [Google Scholar] [CrossRef]
- de Vaux, A.; Laguerre, G.; Diviès, C.; Prévost, H. Enterococcus Asini Sp. Nov. Isolated from the Caecum of Donkeys (Equus asinus). Int. J. Syst. Bacteriol. 1998, 48 Pt 2, 383–387. [Google Scholar] [CrossRef]
- Liu, G.; Bou, G.; Su, S.; Xing, J.; Qu, H.; Zhang, X.; Wang, X.; Zhao, Y.; Dugarjaviin, M. Microbial Diversity within the Digestive Tract Contents of Dezhou Donkeys. PLoS ONE 2019, 14, e0226186. [Google Scholar] [CrossRef] [PubMed]
- Alozie, A.; Köller, K.; Pose, L.; Raftis, M.; Steinhoff, G.; Westphal, B.; Lamprecht, G.; Podbielski, A. Streptococcus bovis infectious endocarditis and occult gastrointestinal neoplasia: Experience with 25 consecutive patients treated surgically. Gut Pathog. 2015, 7, 27. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, Y.; Chen, W.; Lan, T.; Wang, Y.; Wu, Y.; Liao, X.; Mi, J. The Dynamic Changes of Gut Microbiota during the Perinatal Period in Sows. Animals 2020, 10, 2254. [Google Scholar] [CrossRef]
- Paraschou, G.; Cook, J.M.; Priestnall, S.L.; Evans, N.J.; Staton, G.J.; Paterson, G.K.; Winkler, B.; Whitbread, T.J. Treponema spp. Spirochetes and Keratinopathogenic Fungi Isolated from Keratomas in Donkeys. Vet. Pathol. 2023, 60, 190–198. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Q.; Shi, X.; Liu, G.; Wang, C. Integrated Multi-Omics Reveals Novel Microbe-Host Lipid Metabolism and Immune Interactions in the Donkey Hindgut. Front. Immunol. 2022, 13, 1003247. [Google Scholar] [CrossRef]
- Arfuso, F.; Giannetto, C.; Rizzo, M.; Fazio, F.; Giudice, E.; Piccione, G. Serum Levels of Mitochondrial Uncoupling Protein 1, Leptin, and Lipids during Late Pregnancy and the Early Postpartum Period in Mares. Theriogenology 2016, 86, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Weese, J.S.; Holcombe, S.J.; Embertson, R.M.; Kurtz, K.A.; Roessner, H.A.; Jalali, M.; Wismer, S.E. Changes in the Faecal Microbiota of Mares Precede the Development of Post Partum Colic. Equine Vet. J. 2015, 47, 641–649. [Google Scholar] [CrossRef]
- Liu, J.; Hao, W.; He, Z.; Kwek, E.; Zhao, Y.; Zhu, H.; Liang, N.; Ma, K.Y.; Lei, L.; He, W.-S.; et al. Beneficial Effects of Tea Water Extracts on the Body Weight and Gut Microbiota in C57BL/6J Mice Fed with a High-Fat Diet. Food Funct. 2019, 10, 2847–2860. [Google Scholar] [CrossRef]
- Liao, R.; Xie, X.; Lv, Y.; Dai, J.; Lin, Y.; Zhu, L. Ages of Weaning Influence the Gut Microbiota Diversity and Function in Chongming White Goats. Appl. Microbiol. Biotechnol. 2021, 105, 3649–3658. [Google Scholar] [CrossRef]
- Milinovich, G.J.; Burrell, P.C.; Pollitt, C.C.; Klieve, A.V.; Blackall, L.L.; Ouwerkerk, D.; Woodland, E.; Trott, D.J. Microbial Ecology of the Equine Hindgut during Oligofructose-Induced Laminitis. ISME J. 2008, 2, 1089–1100. [Google Scholar] [CrossRef]
- Gu, W.; Zhang, L.; Han, T.; Huang, H.; Chen, J. Dynamic Changes in Gut Microbiome of Ulcerative Colitis: Initial Study from Animal Model. J. Inflamm. Res. 2022, 15, 2631–2647. [Google Scholar] [CrossRef]
- Norman, J.E.; Bollapragada, S.; Yuan, M.; Nelson, S.M. Inflammatory pathways in the mechanism of parturition. BMC Pregnancy Childbirth 2007, 7 (Suppl. S1), S7. [Google Scholar] [CrossRef]
- Waitt, L.H.; Cebra, C.K. Characterization of Hypertriglyceridemia and Response to Treatment with Insulin in Horses, Ponies, and Donkeys: 44 Cases (1995–2005). J. Am. Vet. Med. Assoc. 2009, 234, 915–919. [Google Scholar] [CrossRef]
- Mendoza, F.J.; Toribio, R.E.; Perez-Ecija, A. Metabolic and Endocrine Insights in Donkeys. Animals 2024, 14, 590. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Ji, X.; Chu, X.; Wang, B.; Sun, K.; Wei, H.; Zhang, Y.; Song, Z.; Wen, F. Mulberry Leaf Dietary Supplementation Can Improve the Lipo-Nutritional Quality of Pork and Regulate Gut Microbiota in Pigs: A Comprehensive Multi-Omics Analysis. Animals 2024, 14, 1233. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, Z.; Wang, X.; Zhang, X.; Zhu, Y.; Guo, J. Parents greater influenced the fecal microbiome and resistome of ibis nestlings than artifical breeding environment contamination. Environ. Res. 2025, 271, 121057. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Park, Y.J.; Kim, Y.-R.; Kim, Y.N.; Ka, S.; Lee, H.Y.; Seong, J.K.; Seok, Y.-J.; Kim, J.B. Alteration of Gut Microbiota by Vancomycin and Bacitracin Improves Insulin Resistance via Glucagon-like Peptide 1 in Diet-Induced Obesity. FASEB J. 2015, 29, 2397–2411. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | |||
|---|---|---|---|
| Item | Corn Stover | Concentrate | Total |
| Dry matter (%) | 88.82 | 88.27 | 88.64 |
| Crude protein (%) | 9.02 | 18.77 | 15.13 |
| Crude fiber (%) | 32.30 | 5.81 | 20.64 |
| ADF (%) | 39.33 | 11.82 | 25.68 |
| NDF (%) | 63.78 | 23.51 | 48.43 |
| Ether extract (%) | 1.43 | 4.13 | 2.78 |
| Calcium (%) | 0.31 | 1.13 | 0.95 |
| Phosphorus (%) | 0.10 | 0.81 | 0.52 |
| Magnesium (%) | 3.02 | 0.55 | 2.08 |
| DE (MJ/kg) | 7.53 | 12.23 | 9.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Shao, Y.; Zhou, X.; Li, Z.; Liu, J.; Tang, M.; Yang, Y.; Deng, L. Dynamic Changes in the Gut Microbiota During Peripartum in Jennies. Animals 2025, 15, 1337. https://doi.org/10.3390/ani15091337
Wang X, Shao Y, Zhou X, Li Z, Liu J, Tang M, Yang Y, Deng L. Dynamic Changes in the Gut Microbiota During Peripartum in Jennies. Animals. 2025; 15(9):1337. https://doi.org/10.3390/ani15091337
Chicago/Turabian StyleWang, Xinyue, Yang Shao, Xiaoling Zhou, Zheng Li, Jingze Liu, Mingyao Tang, Yixin Yang, and Liang Deng. 2025. "Dynamic Changes in the Gut Microbiota During Peripartum in Jennies" Animals 15, no. 9: 1337. https://doi.org/10.3390/ani15091337
APA StyleWang, X., Shao, Y., Zhou, X., Li, Z., Liu, J., Tang, M., Yang, Y., & Deng, L. (2025). Dynamic Changes in the Gut Microbiota During Peripartum in Jennies. Animals, 15(9), 1337. https://doi.org/10.3390/ani15091337