
Analysis of Expression Profiles of CircRNA and MiRNA in Oviduct during the Follicular and Luteal Phases of Sheep with Two Fecundity (FecB Gene) Genotypes

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Processing and Sample Acquirement
2.2. RNA Extraction, Library Construction and RNA-Seq
2.3. Identification and Differential Expression Analysis of ciriRNA
2.4. Identification and Differential Expression Analysis of miRNA
2.5. GO and KEGG Analyses of Host Genes of DE circRNAs and Predicted Target Genes of DE miRNAs
2.6. Validation of the Expression of circRNAs and miRNAs
3. Results
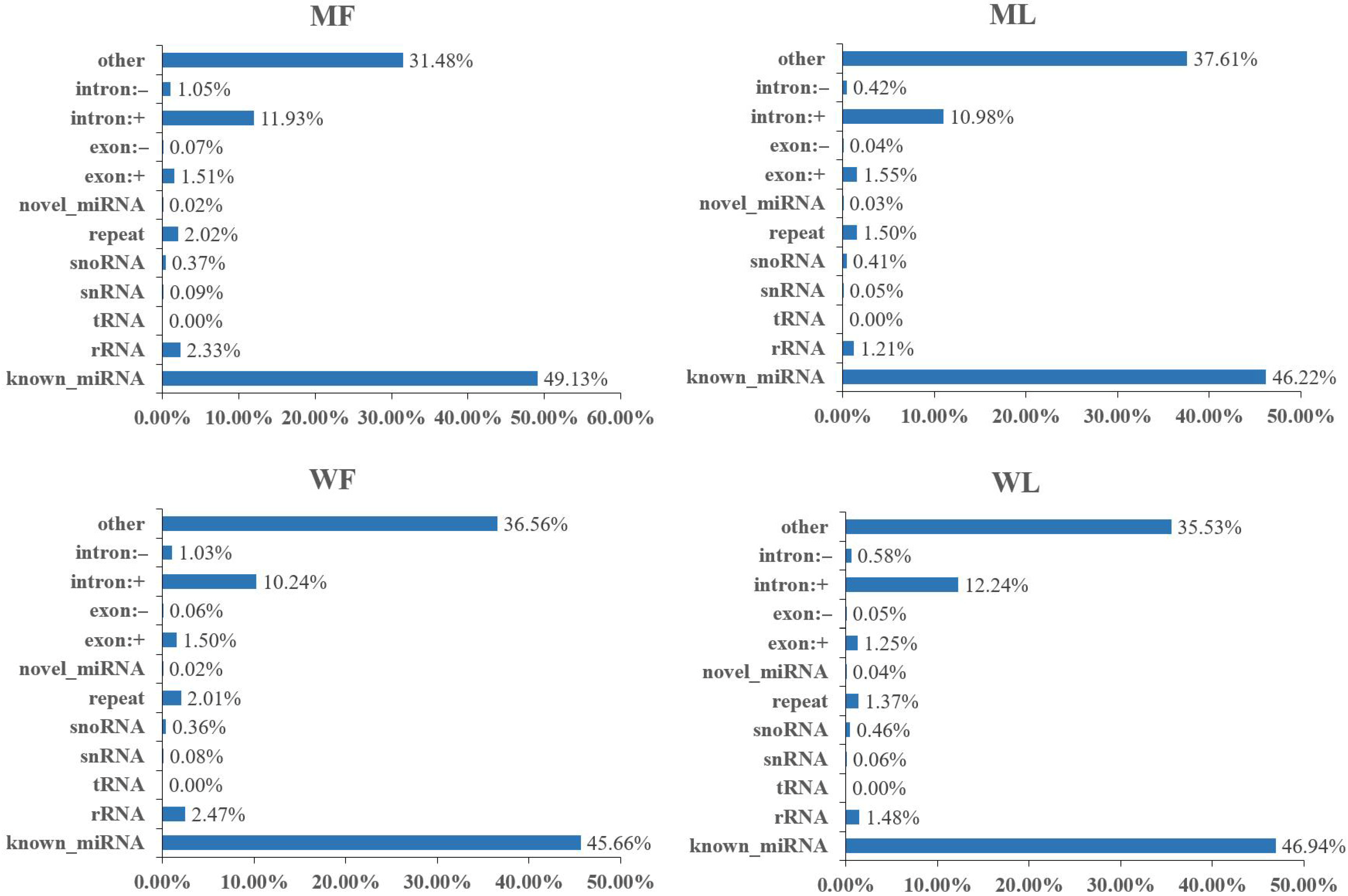
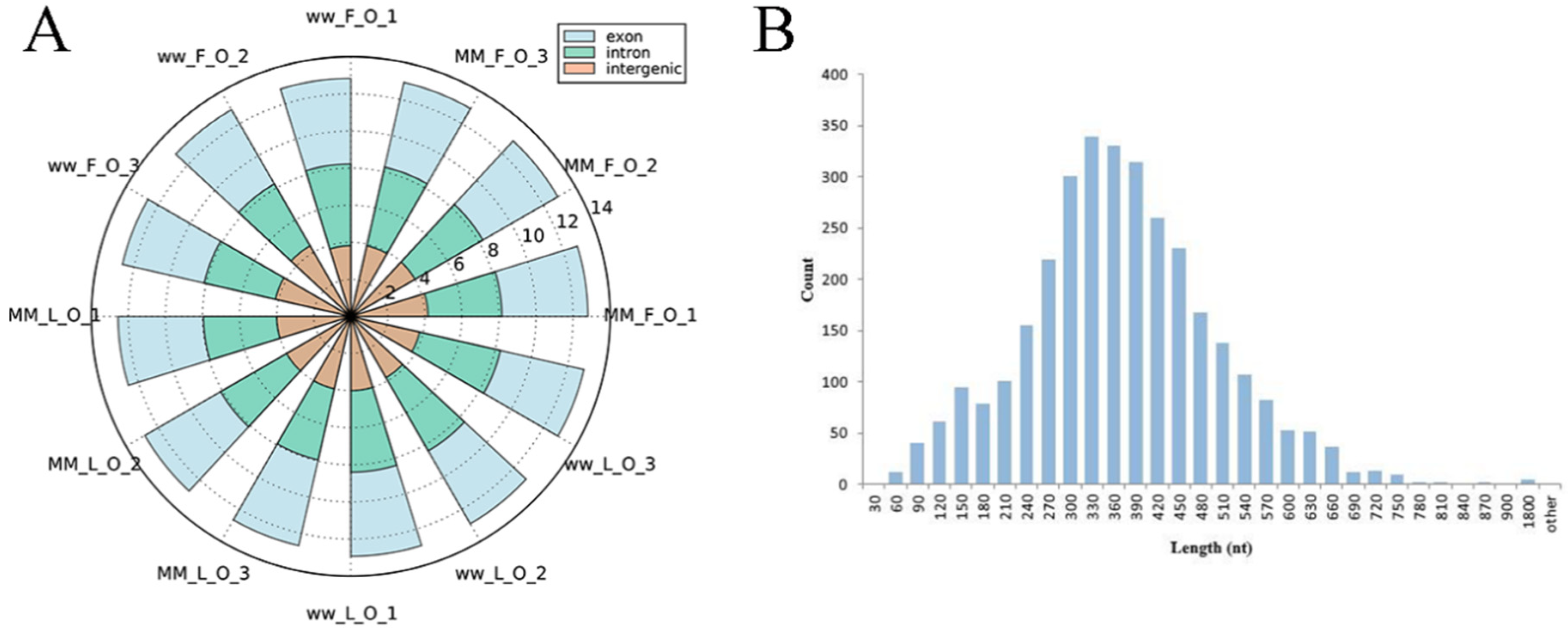
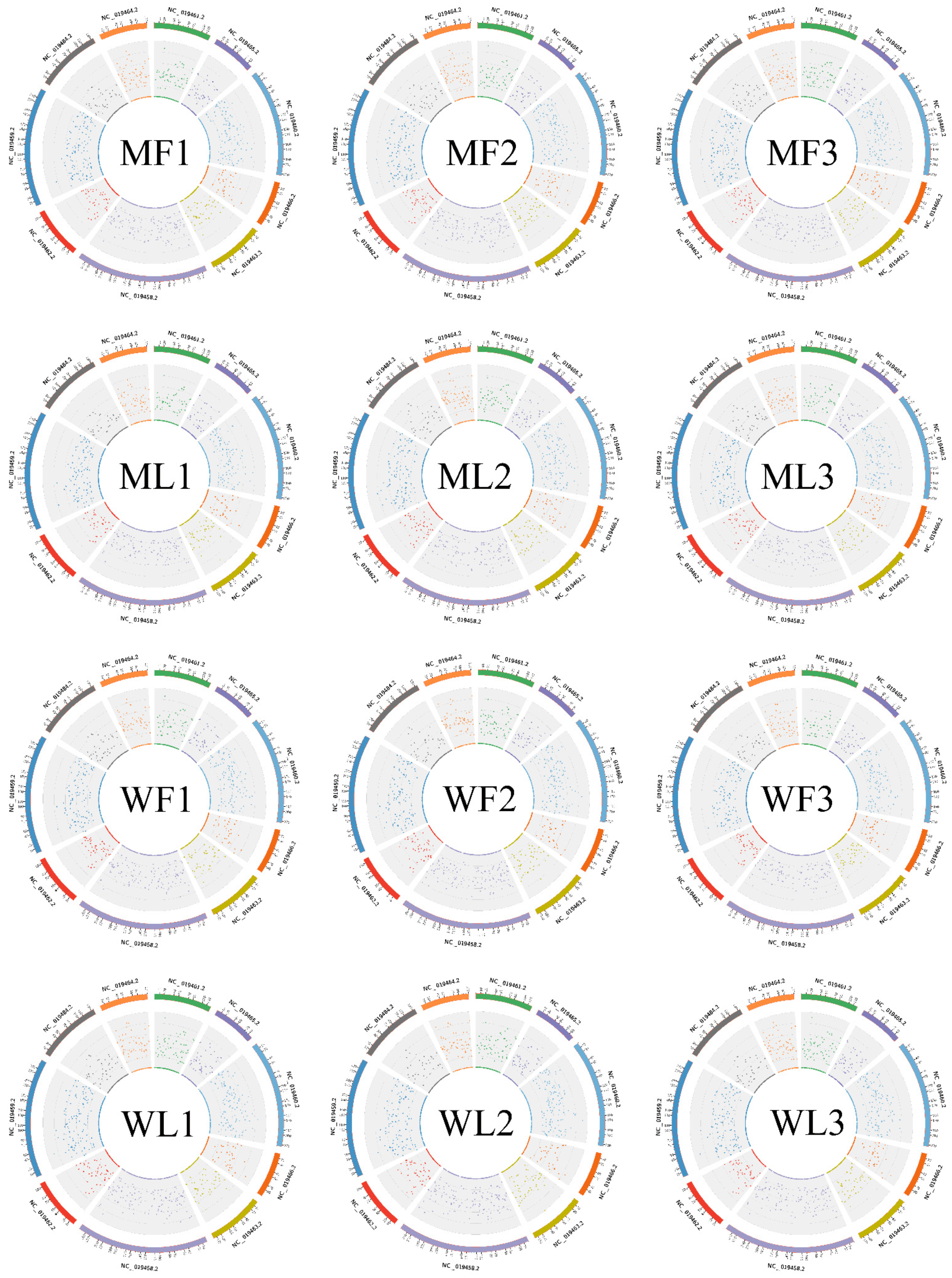
3.1. Overview of circRNA Profiles of Small Tail Han Sheep Oviduct
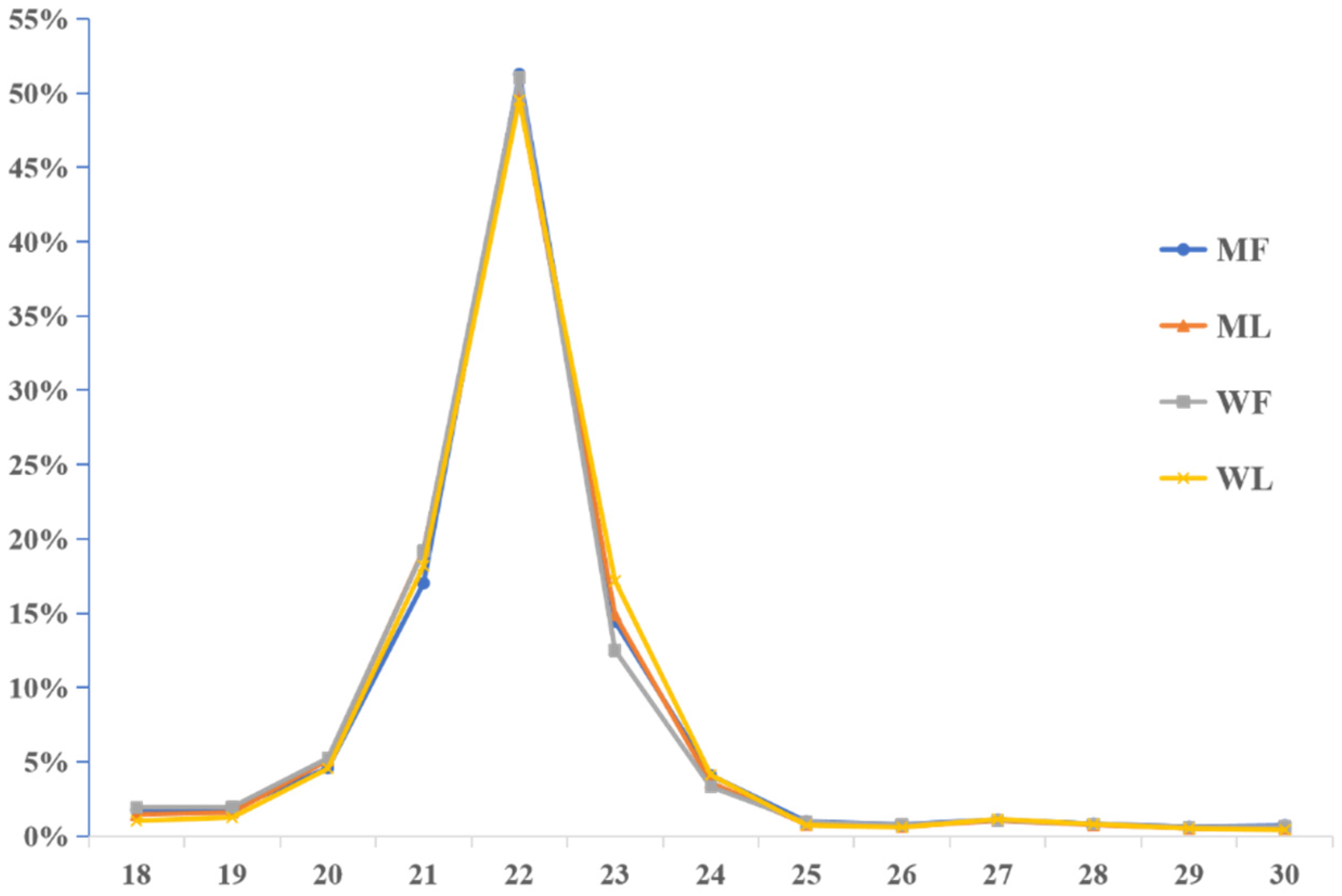
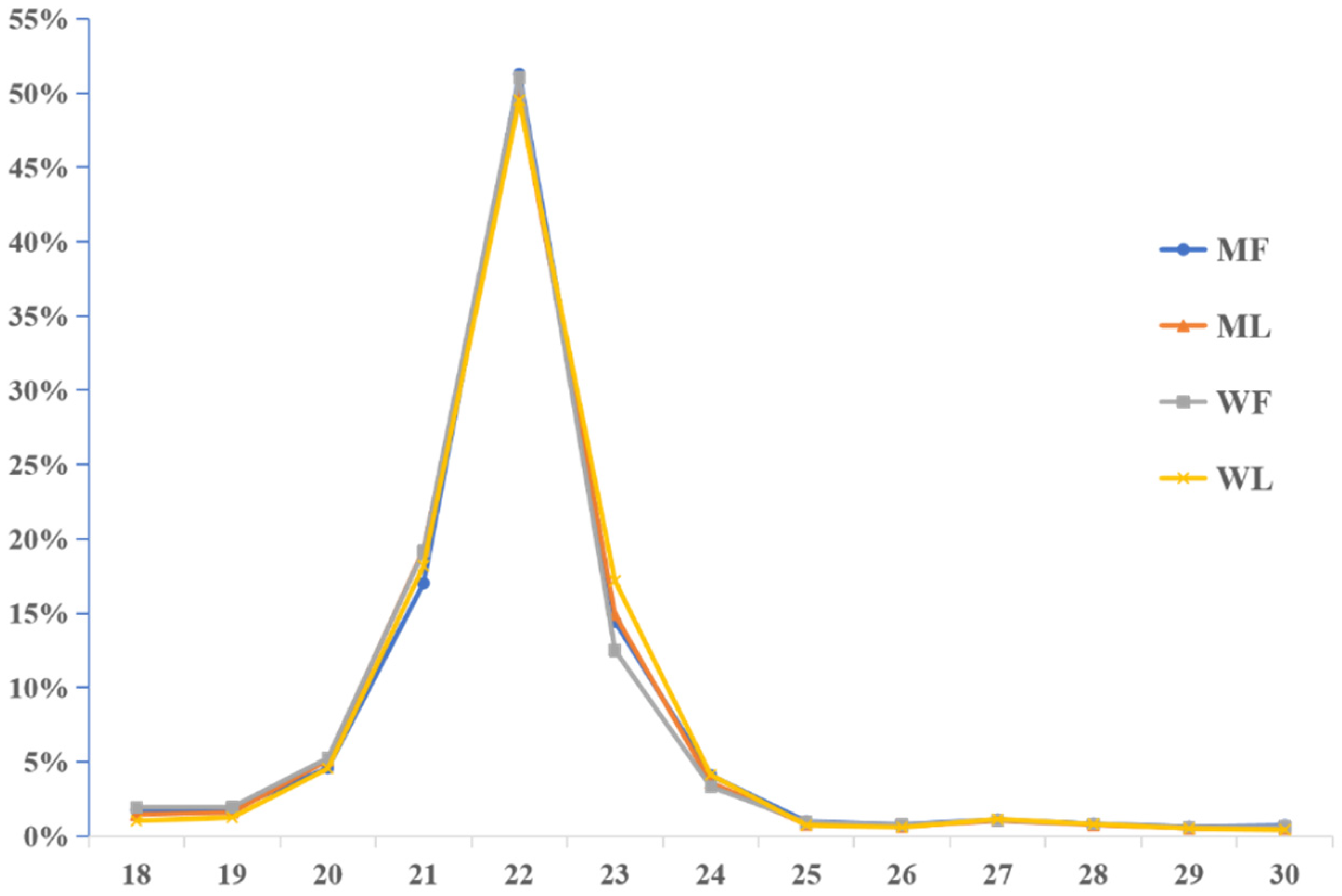
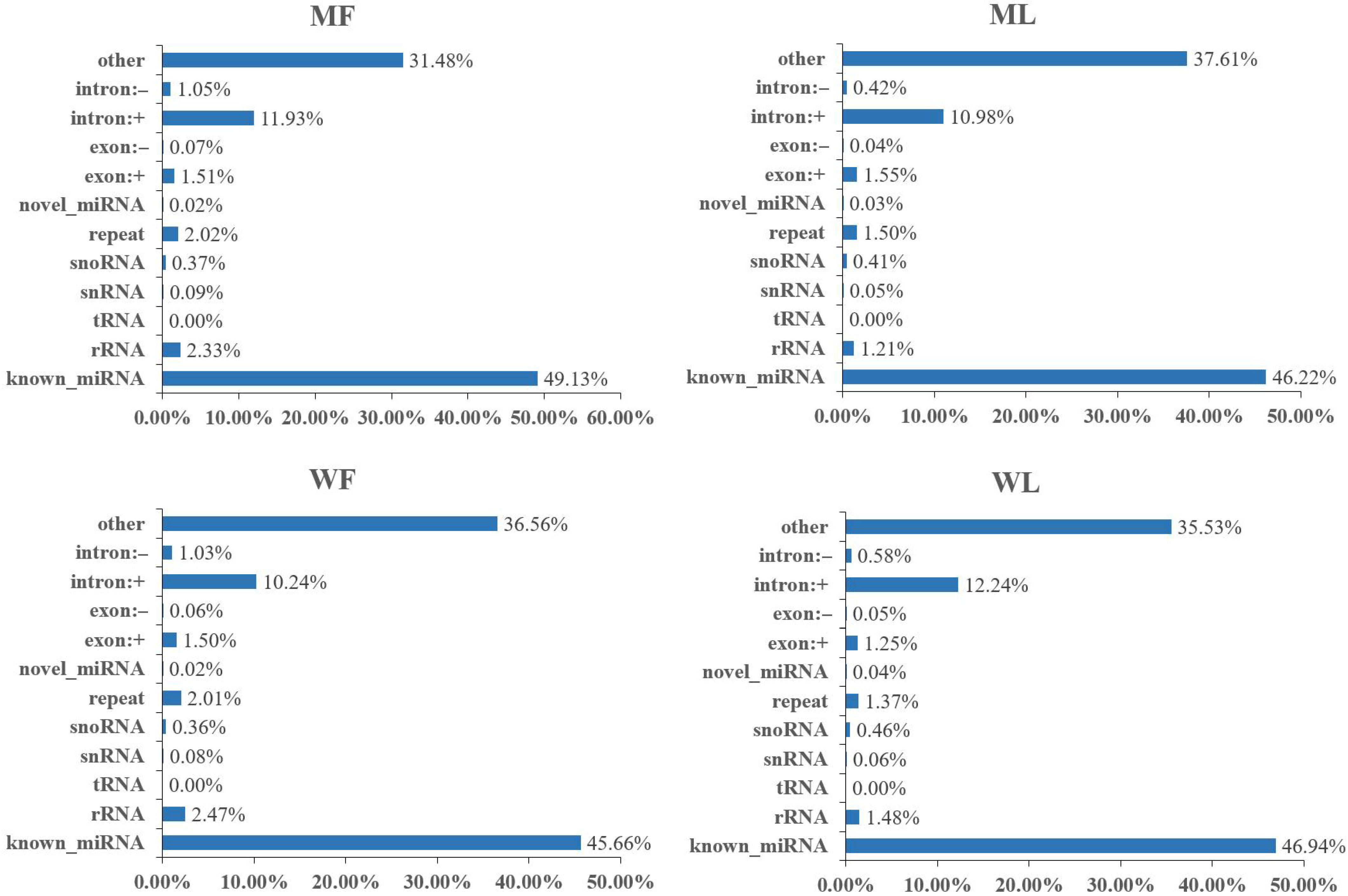
3.2. Overview of miRNA Profiles of Small Tail Han Sheep Oviduct
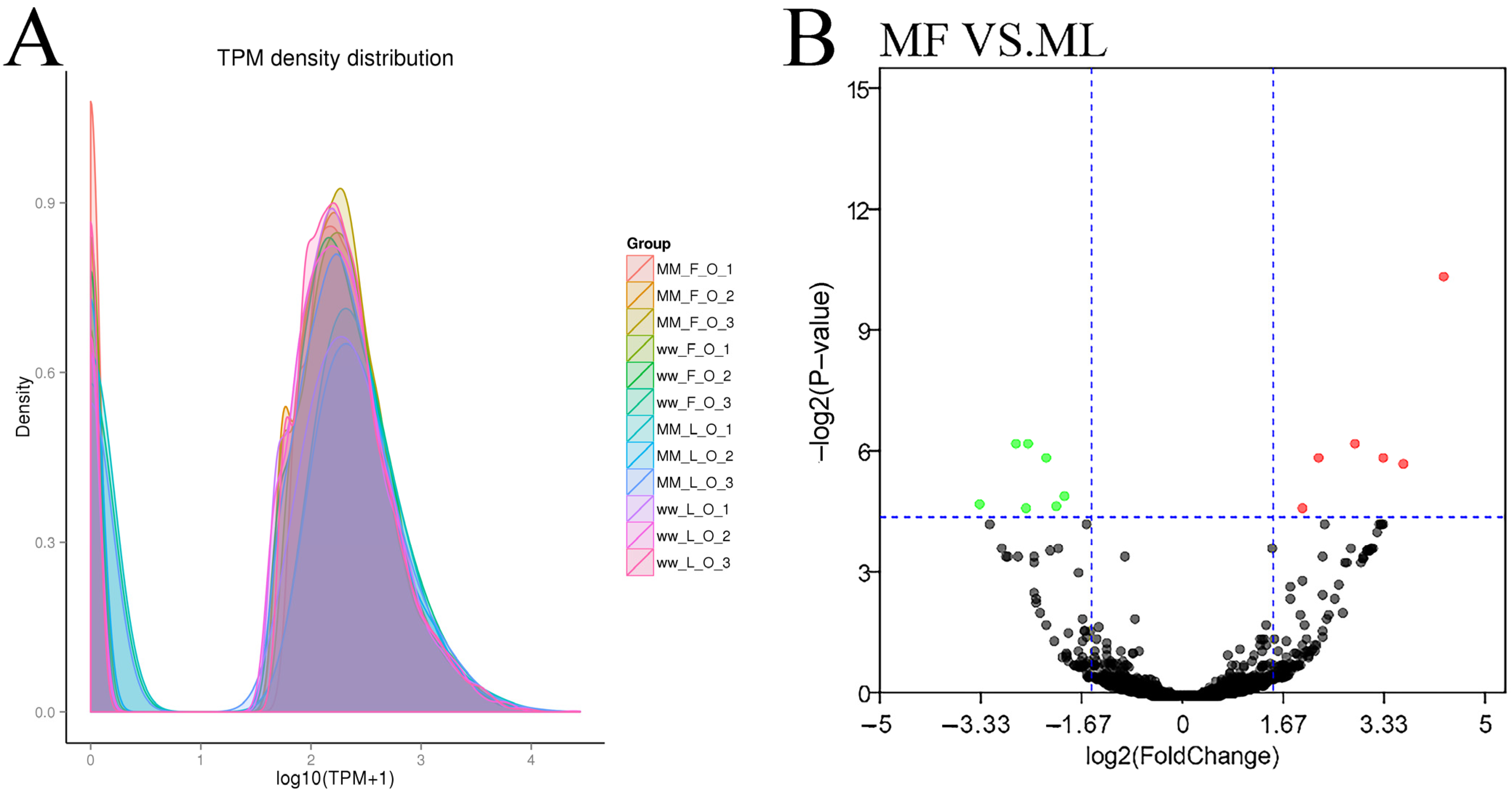
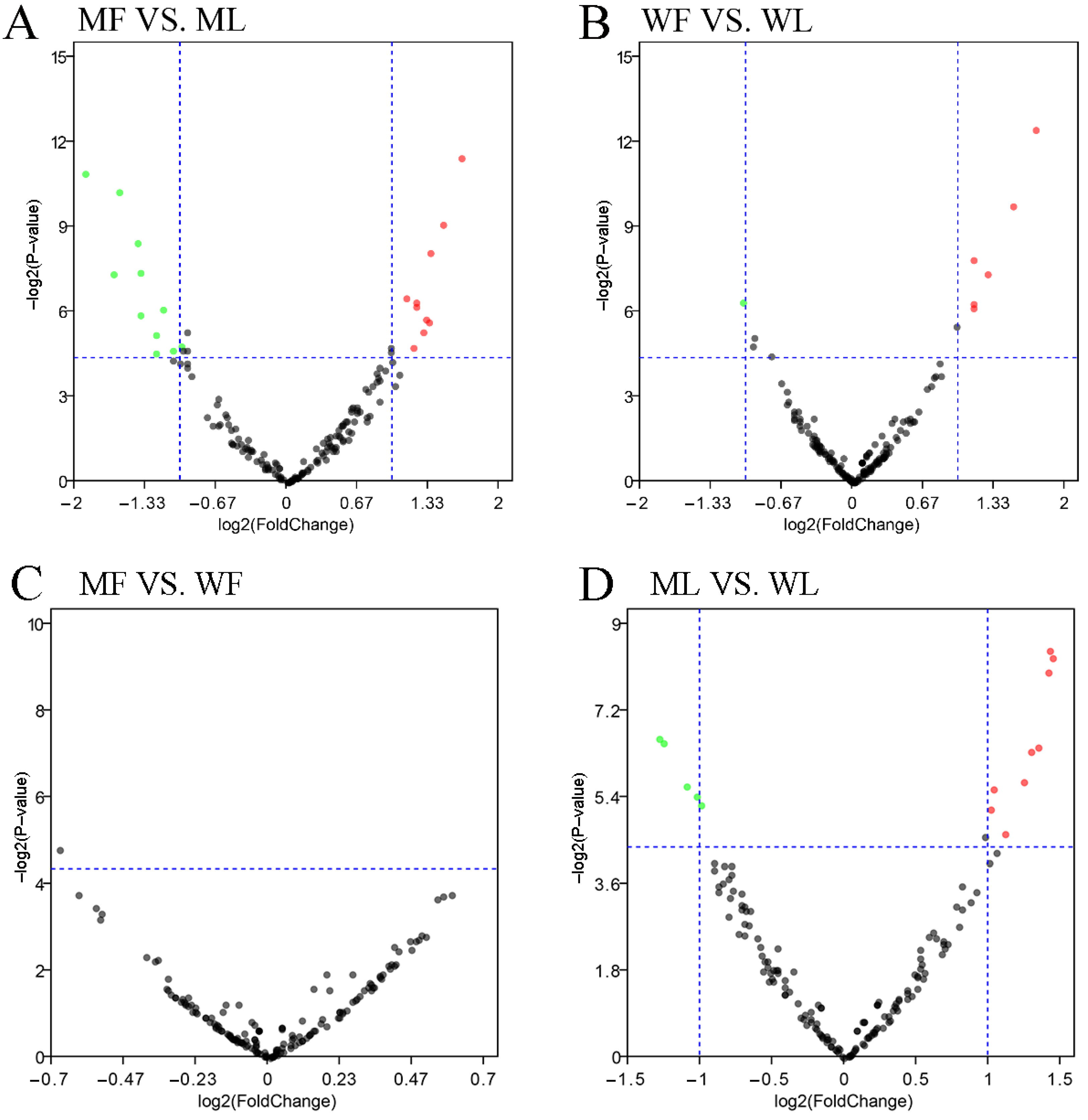
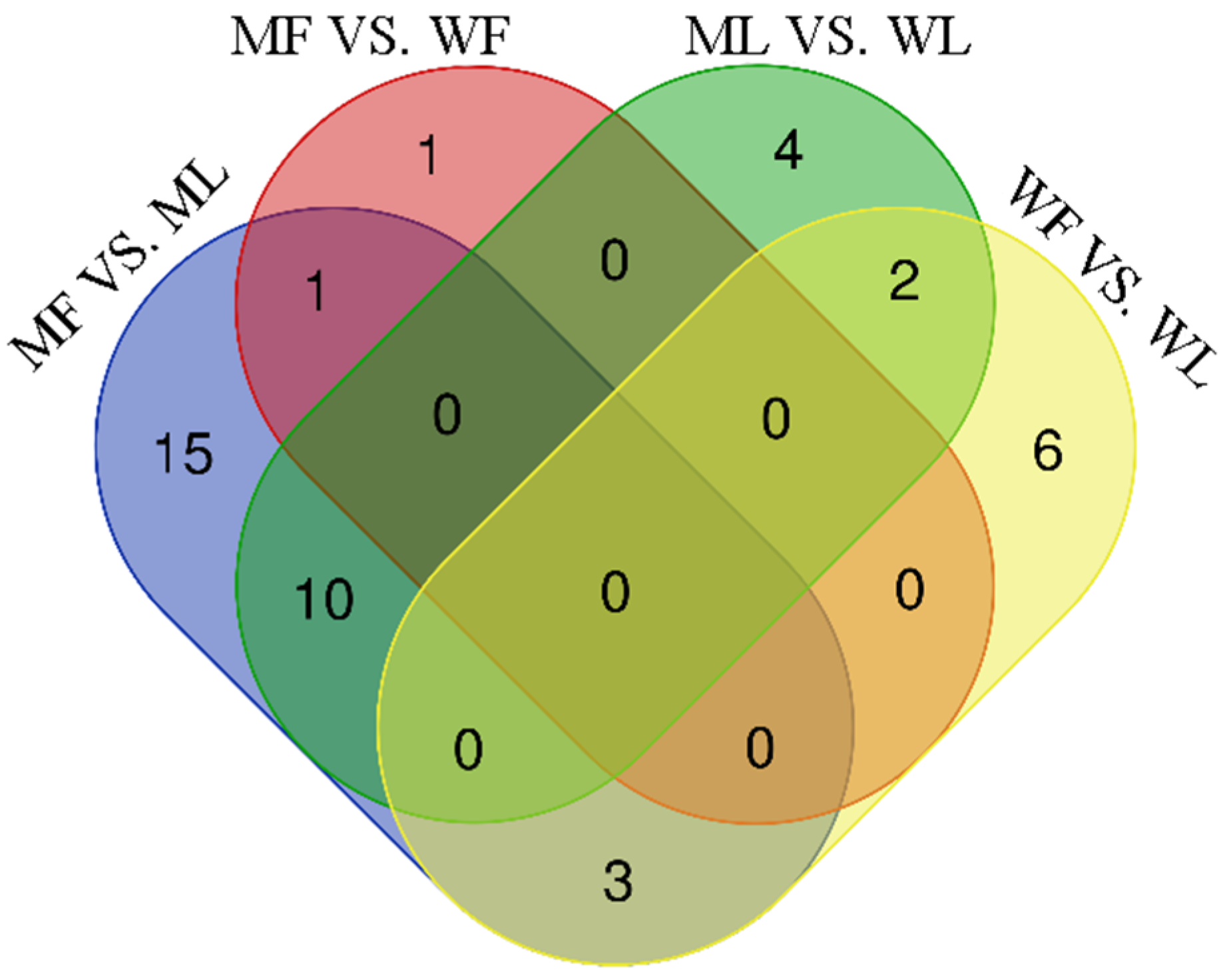
3.3. Differential Expression Analysis of circRNAs
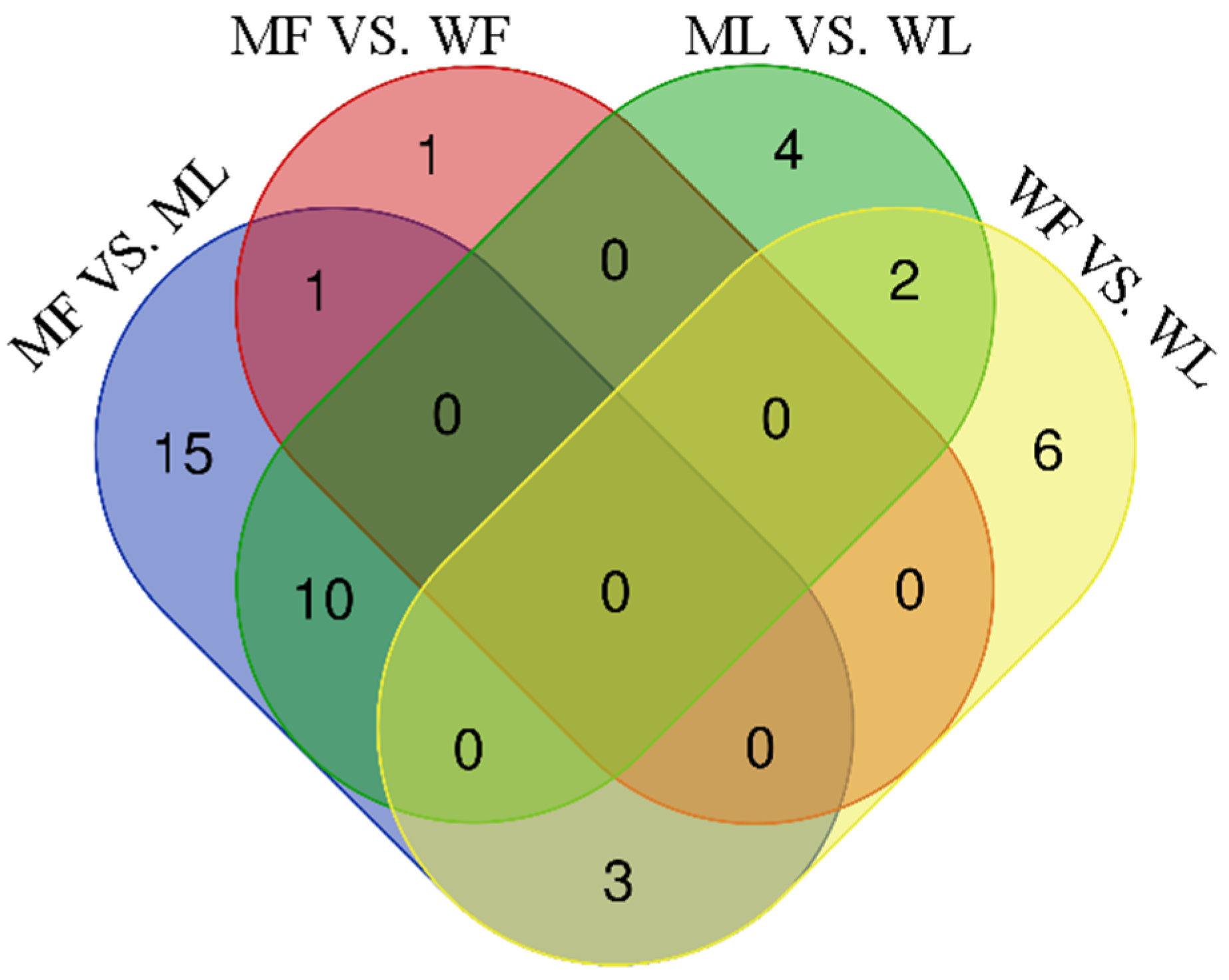
3.4. Differential Expression Analysis of miRNAs
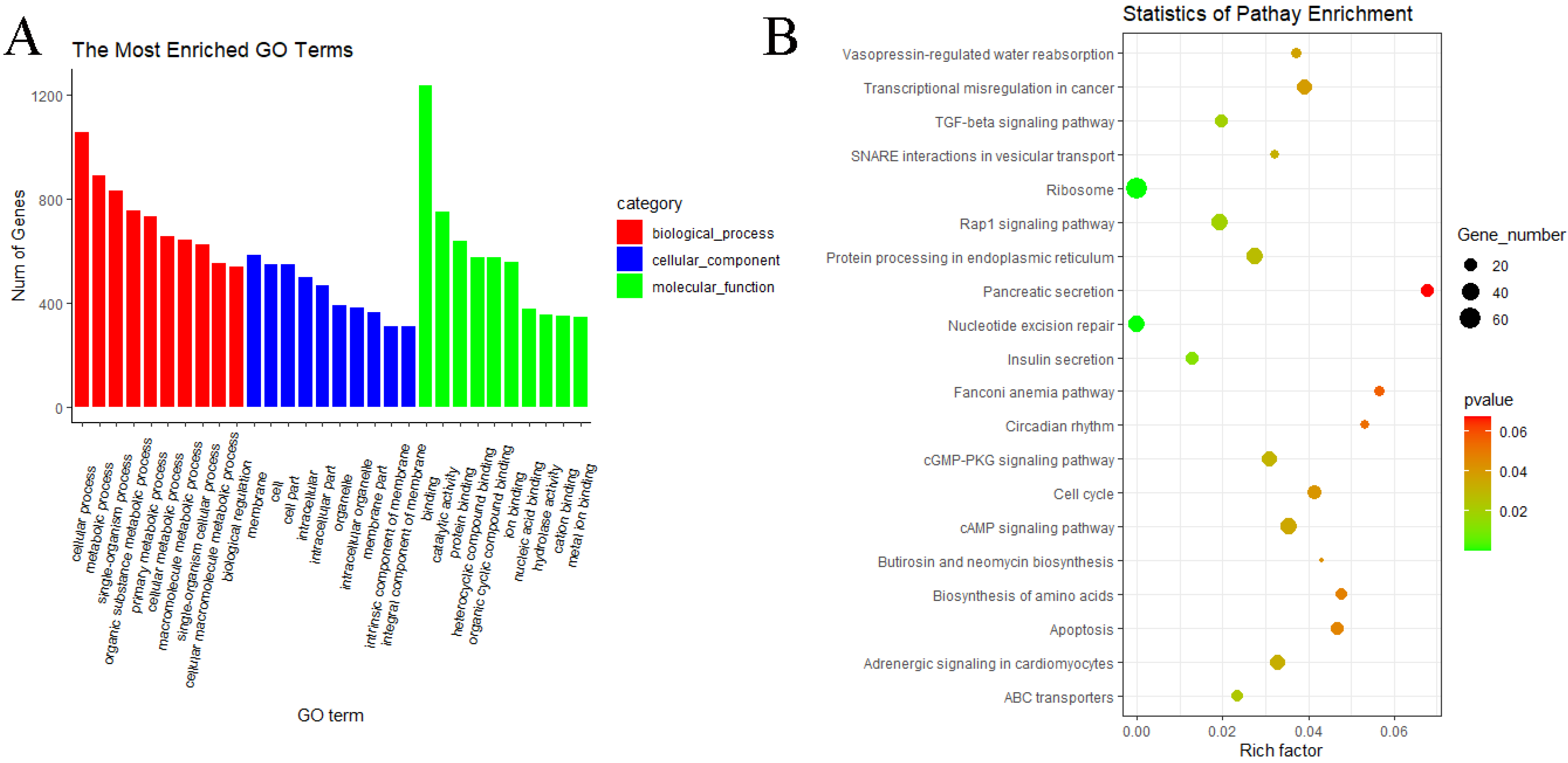
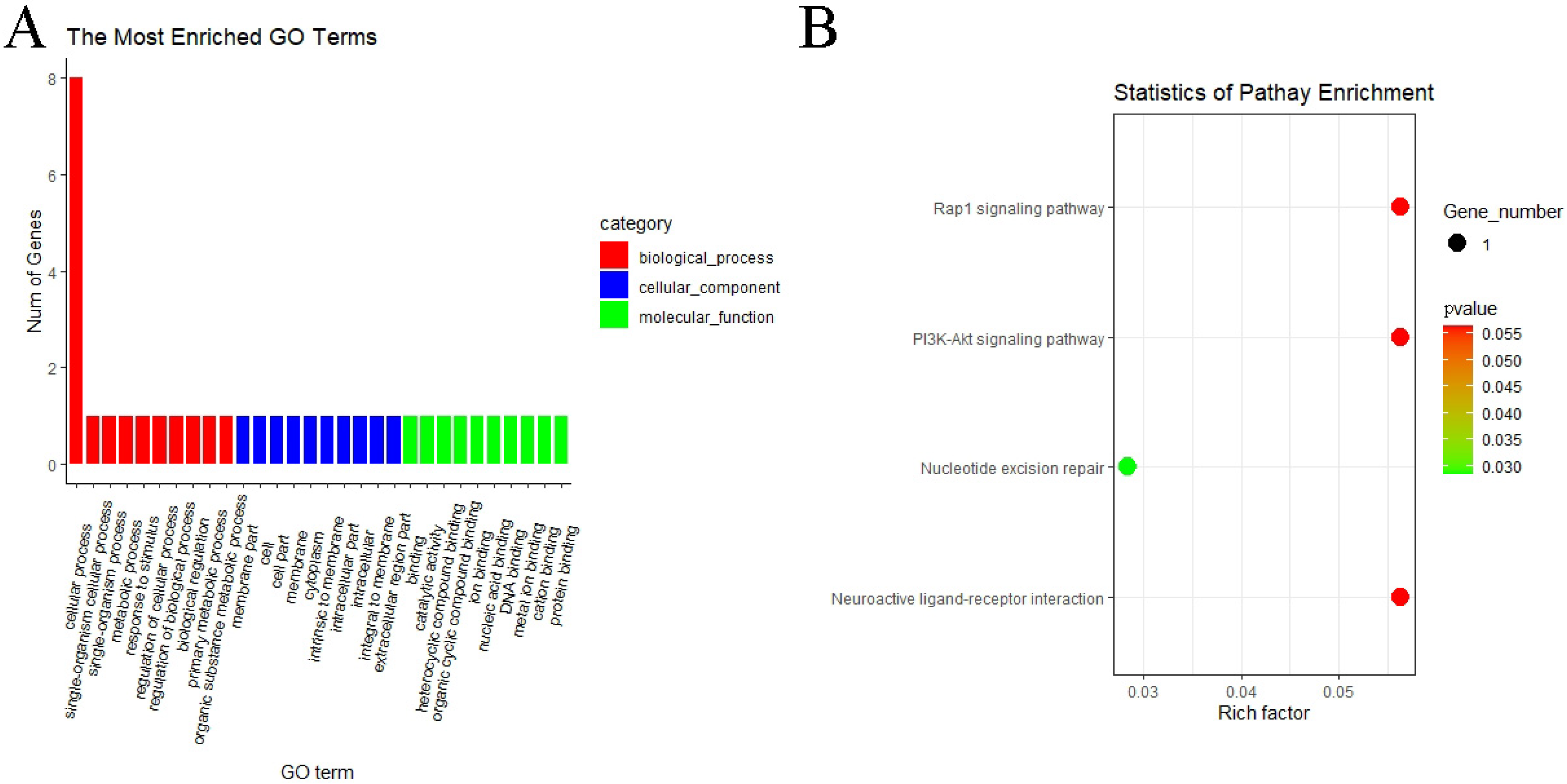
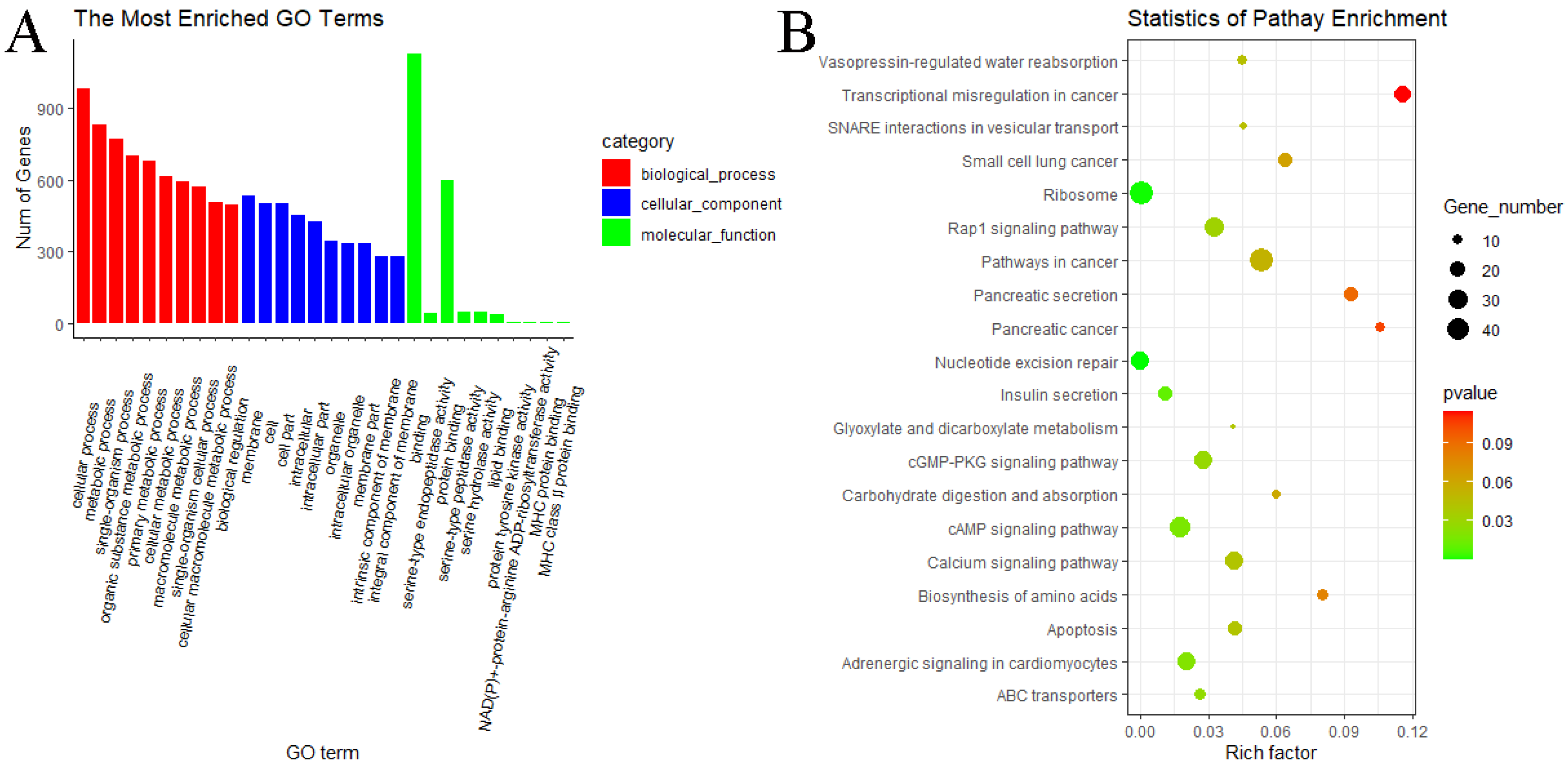
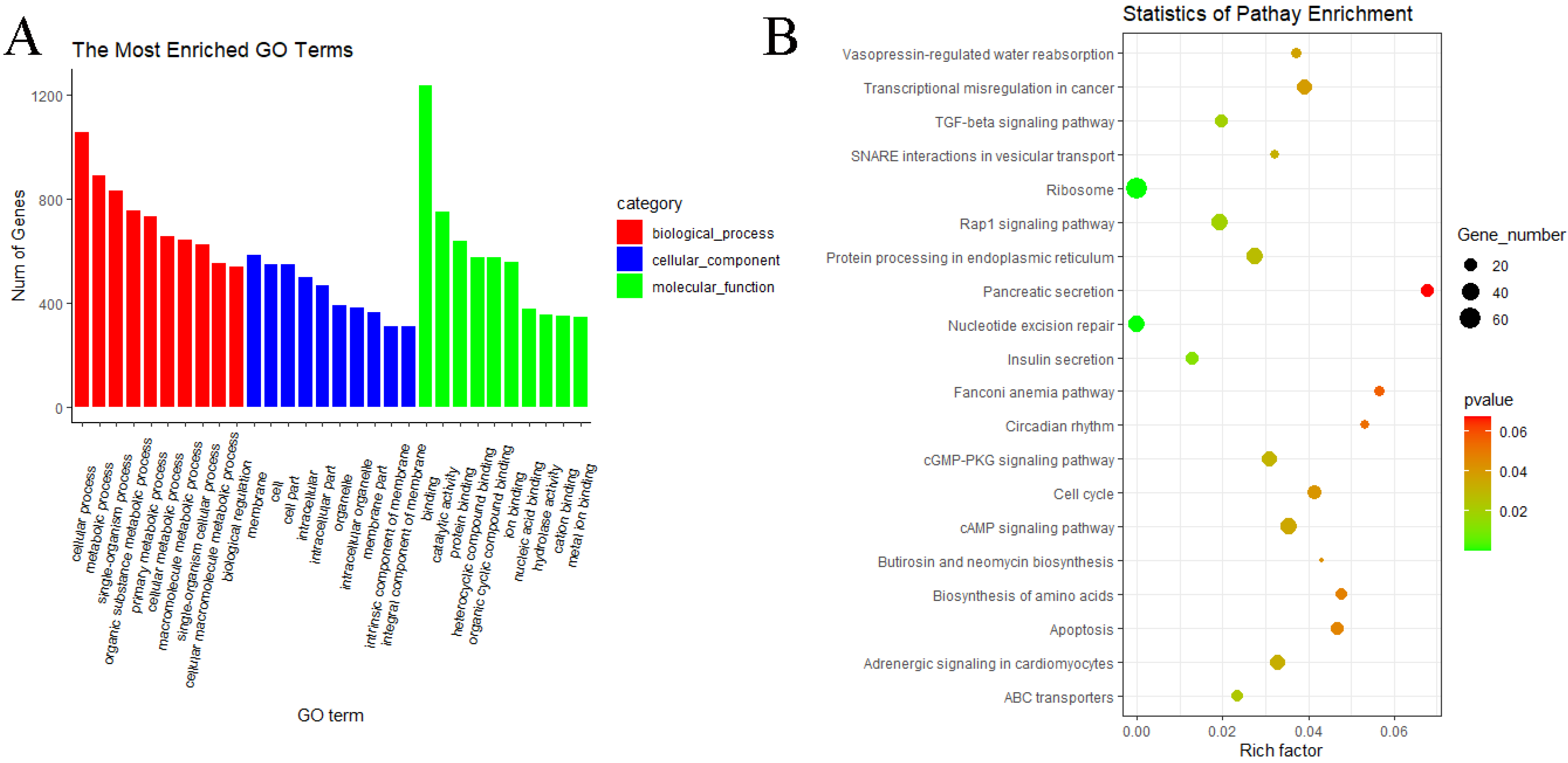
3.5. GO and KEGG Pathway Enrichment Analyses of circRNAs
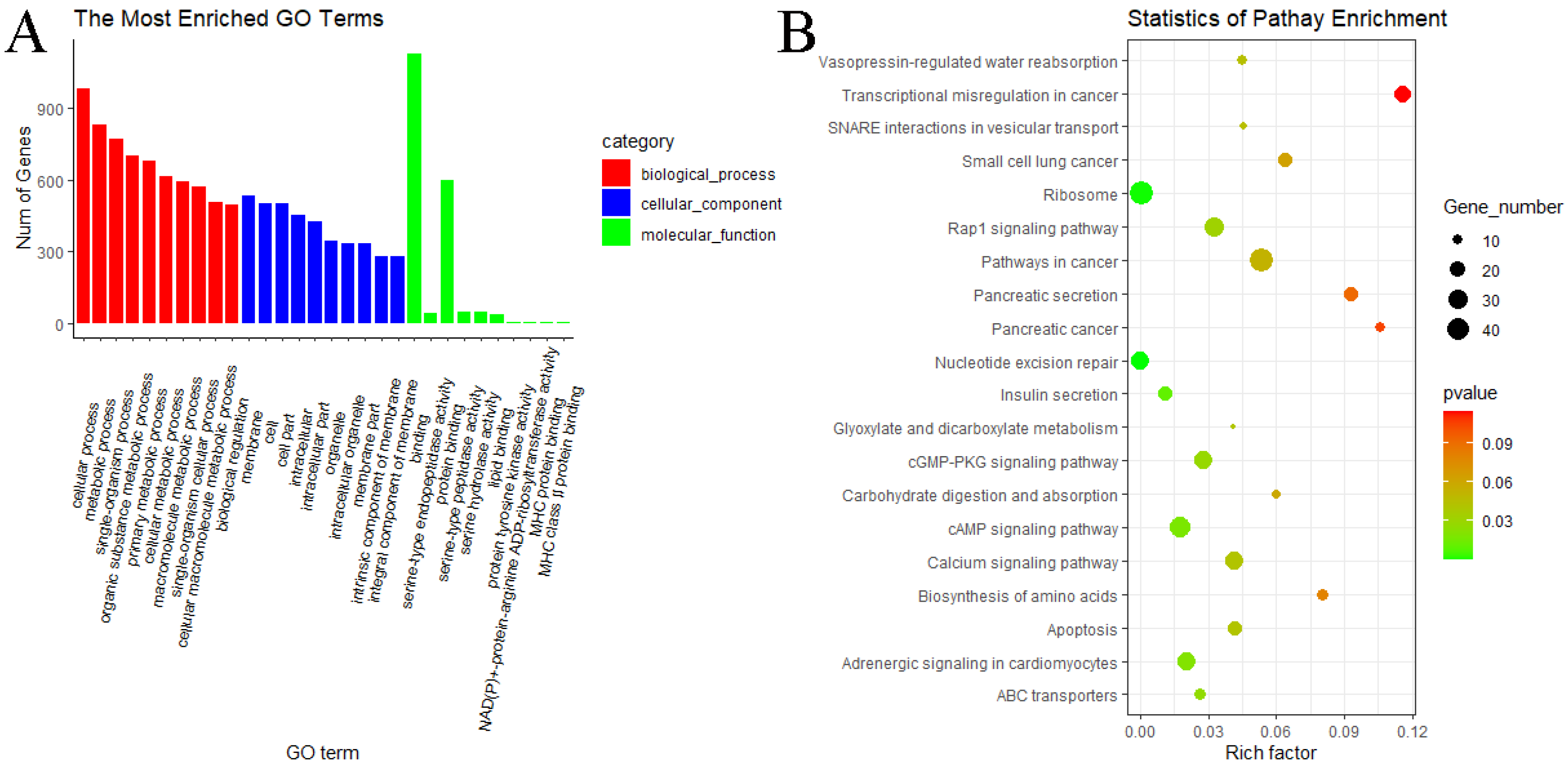
3.6. GO and KEGG Pathway Enrichment Analyses of miRNAs
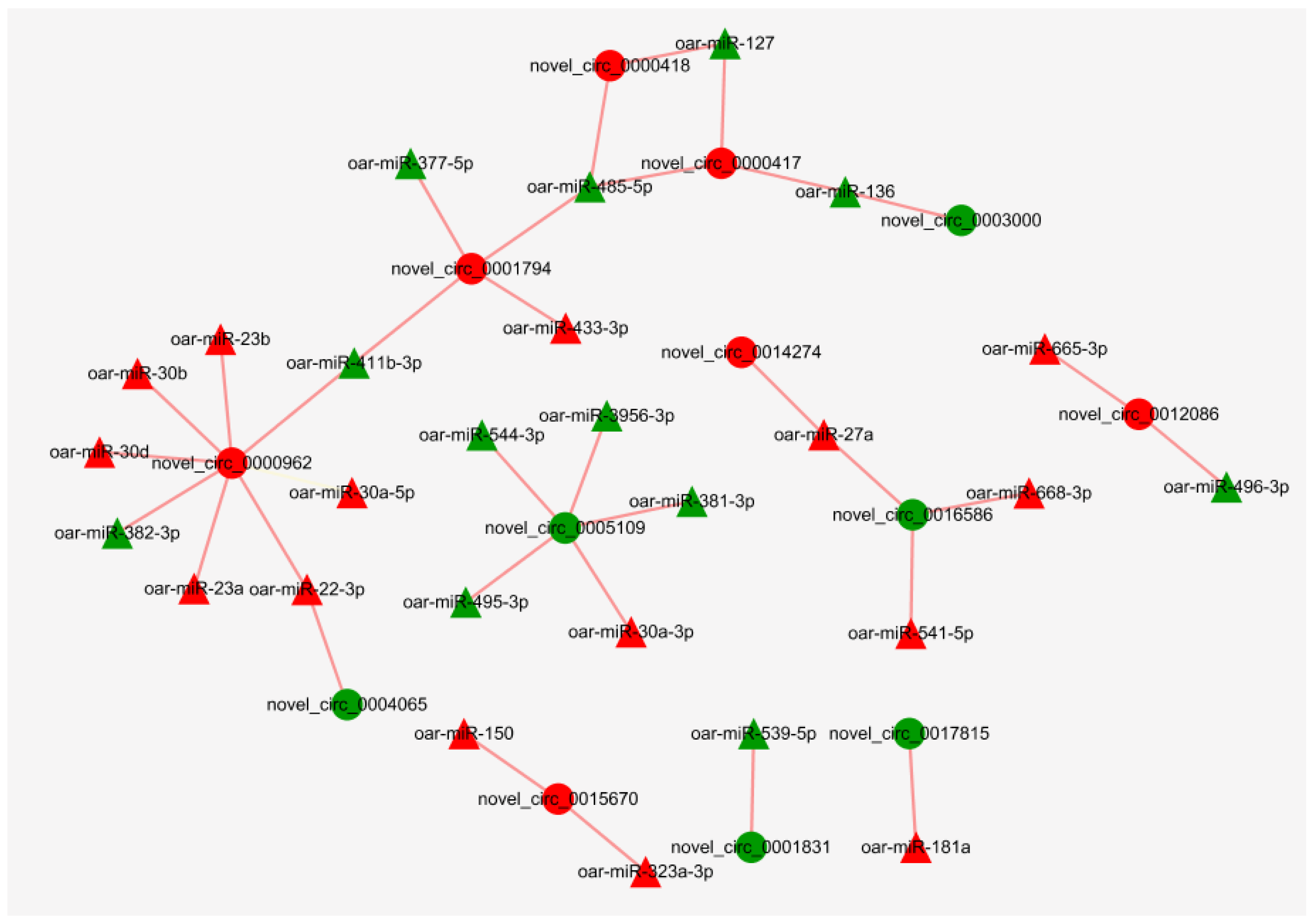
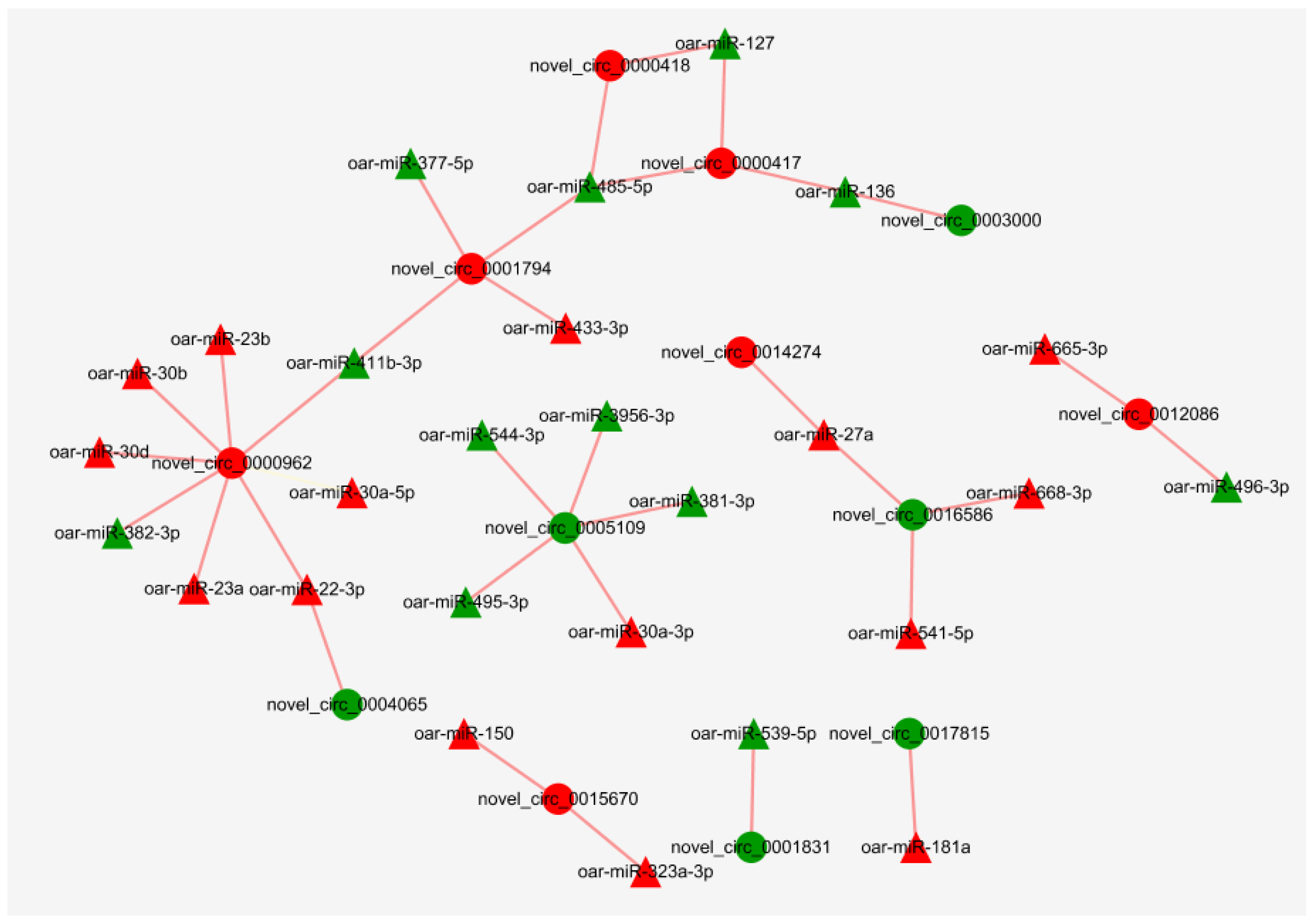
3.7. Regulatory Networks of miRNAs and circRNAs
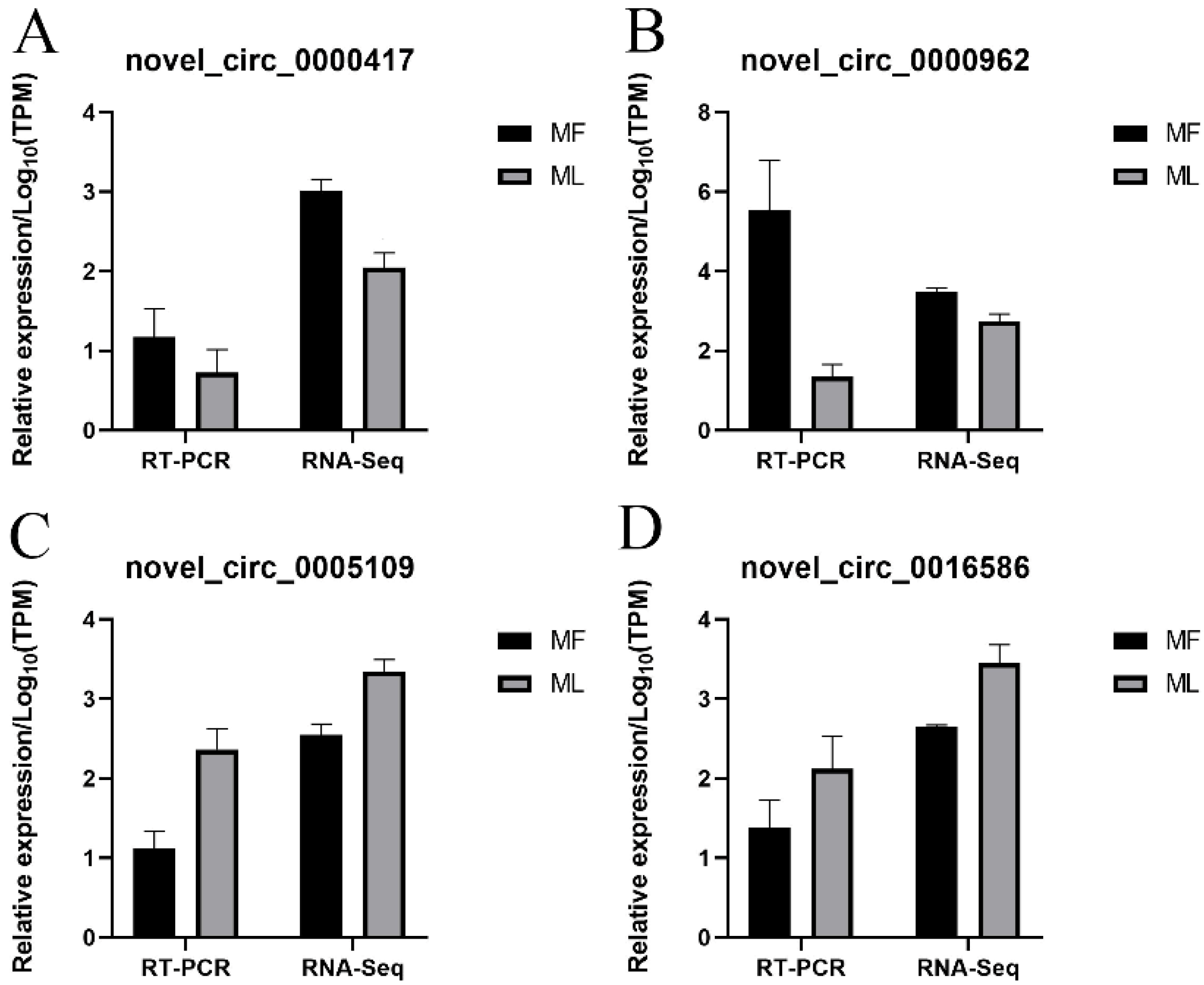
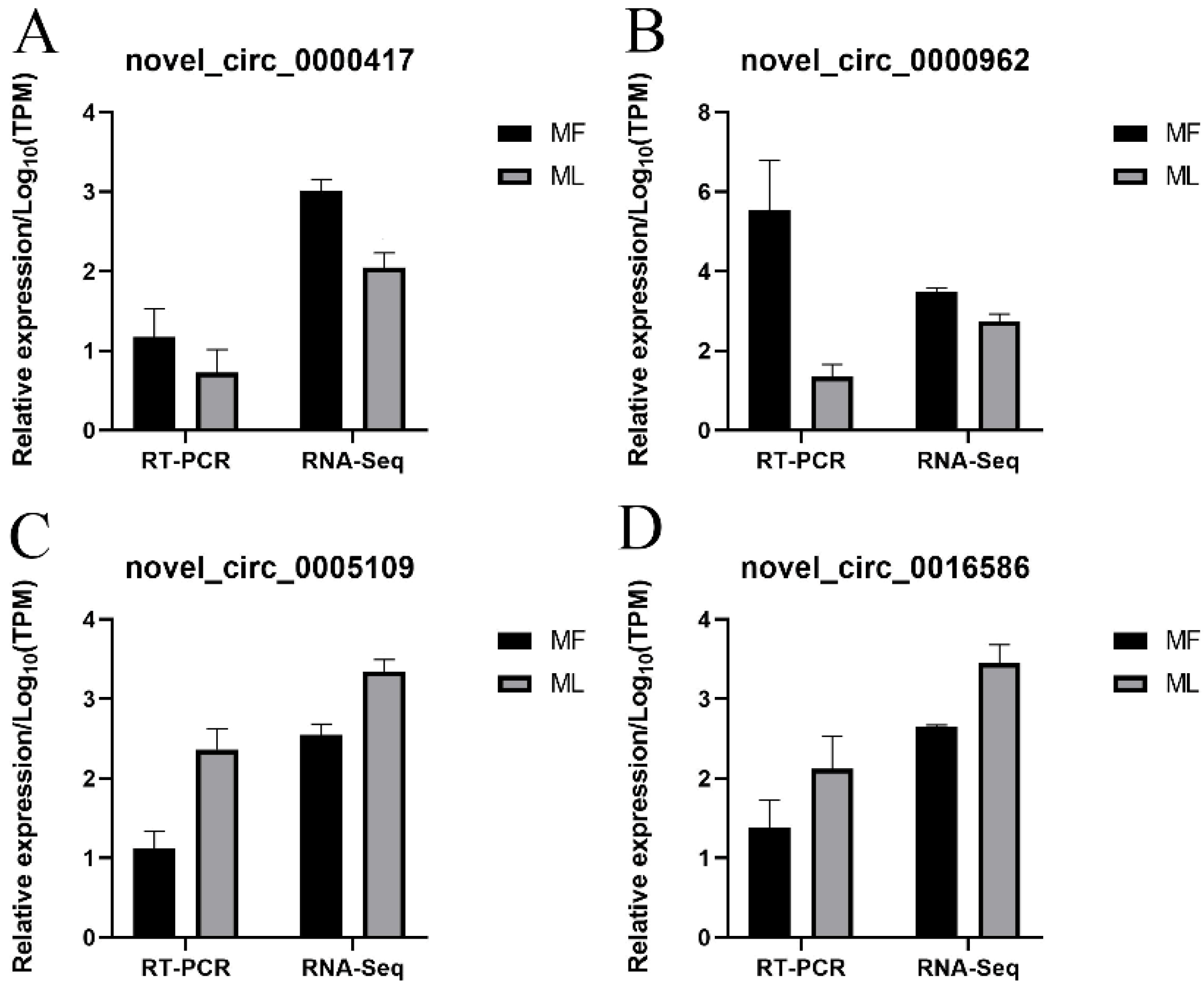
3.8. Validation of circRNA Expression
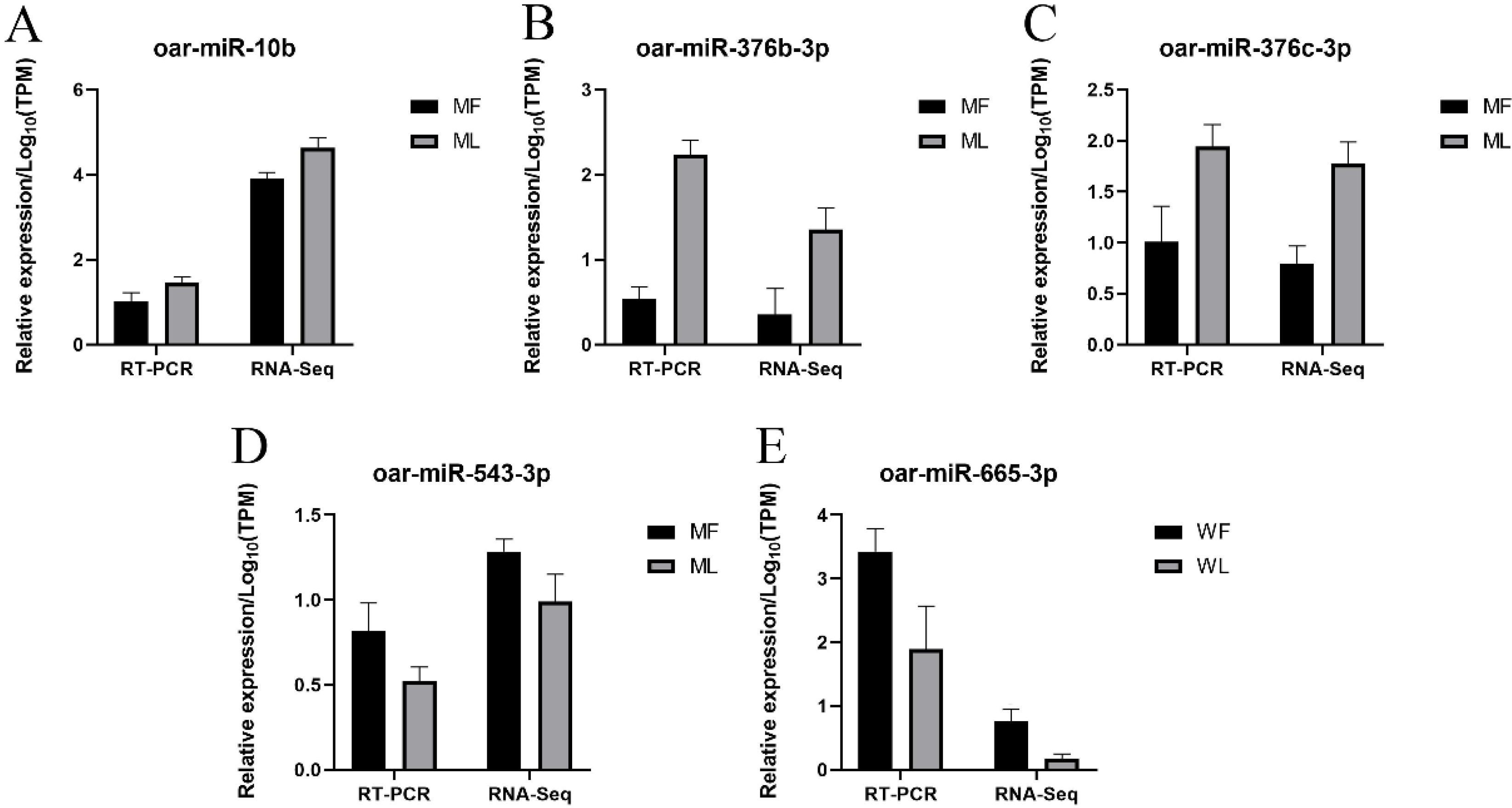
3.9. Validation of miRNA Expression
4. Discussion
4.1. Transcriptomic Profiles
4.2. Differential Expression Analysis and Functional Analysis of circRNAs
4.3. Differential Expression Analysis and Functional Analysis of miRNAs
4.4. Regulatory Networks of miRNAs and circRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian proteomic study reveals the possible molecular mechanism for hyperprolificacy of Small Tail Han sheep. Sci. Rep. 2016, 6, 27606. [Google Scholar] [CrossRef]
- Di, R.; Chu, M.X.; Li, Y.L.; Zhang, L.; Fang, L.; Feng, T.; Cao, G.L.; Chen, H.Q.; Li, X.W. Predictive potential of microsatellite markers on heterosis of fecundity in crossbred sheep. Mol. Biol. Rep. 2012, 39, 2761–2766. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.J.; Macdougall, C.; Macdougall, C.; Campbell, B.K.; Mcneilly, A.S.; Baird, D.T. The Booroola (FecB) phenotype is associated with a mutation in the bone morphogenetic receptor type 1 B (BMPR1B) gene. J. Endocrinol. 2001, 169, R1–R6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulsant, P.; Lecerf, F.; Fabre, S.; Schibler, L.; Monget, P.; Lanneluc, I.; Pisselet, C.; Riquet, J.; Monniaux, D.; Callebaut, I.; et al. Mutation in bone morphogenetic protein receptor-IB is associated with increased ovulation rate in Booroola Merino ewes. Proc. Natl. Acad. Sci. USA 2001, 98, 5104–5109. [Google Scholar] [CrossRef] [Green Version]
- Chu, M.; Jia, L.; Zhang, Y.; Jin, M.; Chen, H.; Fang, L.; Di, R.; Cao, G.; Feng, T.; Tang, Q.; et al. Polymorphisms of coding region of BMPR-IB gene and their relationship with litter size in sheep. Mol. Biol. Rep. 2011, 38, 4071–4076. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian transcriptomic study reveals the differential regulation of miRNAs and lncRNAs related to fecundity in different sheep. Sci. Rep. 2016, 6, 35299. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Qin, X. Genome-wide transcriptome analysis in the ovaries of two goats identifies differentially expressed genes related to fecundity. Gene 2016, 582, 69–76. [Google Scholar] [CrossRef]
- Maillo, V.; Lopera-Vasquez, R.; Hamdi, M.; Gutierrez-Adan, A.; Lonergan, P.; Rizos, D. Maternal-embryo interaction in the bovine oviduct: Evidence from in vivo and in vitro studies. Theriogenology 2016, 86, 443–450. [Google Scholar] [CrossRef]
- Alminana, C.; Corbin, E.; Tsikis, G.; Alcantara-Neto, A.S.; Labas, V.; Reynaud, K.; Galio, L.; Uzbekov, R.; Garanina, A.S.; Druart, X.; et al. Oviduct extracellular vesicles protein content and their role during oviduct-embryo cross-talk. Reproduction 2017, 154, 153–168. [Google Scholar] [CrossRef]
- Memili, E.; First, N.L. Zygotic and embryonic gene expression in cow: A review of timing and mechanisms of early gene expression as compared with other species. Zygote 2000, 8, 87–96. [Google Scholar] [CrossRef]
- Graf, A.; Krebs, S.; Zakhartchenko, V.; Schwalb, B.; Blum, H.; Wolf, E. Fine mapping of genome activation in bovine embryos by RNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 4139–4144. [Google Scholar] [CrossRef] [Green Version]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, X.; Che, S.; Cui, J.; Liu, Y.; An, X.; Cao, B.; Song, Y. CircRNA-9119 regulates the expression of prostaglandin-endoperoxide synthase 2 (PTGS2) by sponging miR-26a in the endometrial epithelial cells of dairy goat. Reprod. Fertil. Dev. 2018, 30, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Ma, Q.; Zhang, X.; Cao, Y.; Yao, Y.; You, S.; Wang, D.; Quan, R.; Hou, X.; et al. Genome-wide analysis of circular RNAs in prenatal and postnatal pituitary glands of sheep. Sci. Rep. 2017, 7, 16143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tang, J.; He, X.; Zhu, M.; Gan, S.; Guo, X.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Comparative transcriptomics identify key hypothalamic circular RNAs that participate in sheep (Ovis aries) reproduction. Animals 2019, 9, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La, Y.; Tang, J.; Di, R.; Wang, X.; Liu, Q.; Zhang, L.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Differential expression of circular RNAs in polytocous and monotocous uterus during the reproductive cycle of sheep. Animals 2019, 9, 797. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Lu, T.; Zhao, Z.; Liu, G.; Lian, Z.; Guo, Y.; Sun, B.; Liu, D.; Li, Y. Comprehensive analysis of mRNAs and miRNAs in the ovarian follicles of uniparous and multiple goats at estrus phase. BMC Genom. 2020, 21, 267. [Google Scholar] [CrossRef]
- Donadeu, F.X.; Schauer, S.N.; Sontakke, S.D. Involvement of miRNAs in ovarian follicular and luteal development. J. Endocrinol. 2012, 215, 323–334. [Google Scholar] [CrossRef]
- Sun, L.; Lu, S.; Bai, M.; Xiang, L.; Li, J.; Jia, C.; Jiang, H. Integrative microRNA-mRNA analysis of muscle tissues in qianhua mutton merino and small tail han sheep reveals key roles for oar-miR-655-3p and oar-miR-381-5p. DNA Cell Biol. 2019, 38, 423–435. [Google Scholar] [CrossRef]
- Corradi, E.; Dalla, C.I.; Gavoci, A.; Iyer, A.; Roccuzzo, M.; Otto, T.A.; Oliani, E.; Bridi, S.; Strohbuecker, S.; Santos-Rodriguez, G.; et al. Axonal precursor miRNAs hitchhike on endosomes and locally regulate the development of neural circuits. EMBO J. 2020, 39, e102513. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Di, R.; Liu, Q.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Chu, M.; Hu, W. Integrated hypothalamic transcriptome profiling reveals the reproductive roles of mRNAs and miRNAs in sheep. Front. Genet. 2019, 10, 1296. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtgast, E.J.; Sima, V.M.; Bertels, K.; Al-Ars, Z. Hardware acceleration of BWA-MEM genomic short read mapping for longer read lengths. Comput. Biol. Chem. 2018, 75, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated profiling of microRNAs and mRNAs: MicroRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- La, Y.; He, X.; Zhang, L.; Di, R.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Comprehensive analysis of differentially expressed profiles of mRNA, lncRNA, and circRNA in the uterus of seasonal reproduction sheep. Genes 2020, 11, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Liu, S.; Tian, F.; Ding, N.; Li, X.; Zhao, K. Comparative transcriptome of reproductive axis in Chinese indigenous sheep with different FecB genotypes and prolificacies. Anim. Reprod. Sci. 2020, 223, 106624. [Google Scholar] [CrossRef]
- Besenfelder, U.; Havlicek, V.; Brem, G. Role of the oviduct in early embryo development. Reprod. Domest. Anim. 2012, 47 (Suppl. 4), 156–163. [Google Scholar] [CrossRef] [Green Version]
- Perez-Cerezales, S.; Ramos-Ibeas, P.; Acuna, O.S.; Aviles, M.; Coy, P.; Rizos, D.; Gutierrez-Adan, A. The oviduct: From sperm selection to the epigenetic landscape of the embryo. Biol. Reprod. 2018, 98, 262–276. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, C.; Wei, J.; Ni, W.; Xu, Y.; Yao, R.; Zhang, M.; Li, H.; Liu, L.; Dang, H.; et al. Comprehensive expression profiling analysis of pituitary indicates that circRNA participates in the regulation of sheep estrus. Genes 2019, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Zhou, H.; Hickford, J.; Gong, H.; Wang, J.; Hu, J.; Liu, X.; Li, S.; Zhao, M.; Luo, Y. Identification and characterization of circular RNA in lactating mammary glands from two breeds of sheep with different milk production profiles using RNA-Seq. Genomics 2020, 112, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Ni, W.; Wang, D.; Hou, X.; Liu, Z.; Cao, Y.; Yao, Y.; Zhang, X.; Hu, S. Identification and comparison of microRNAs in pituitary gland during prenatal and postnatal stages of sheep by deep sequencing. J. Genet. 2018, 97, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Chen, H.Y.; Jia, B.; Han, G.H.; Zhang, Y.S.; Zeng, X.C. Characterization and expression analysis of microRNAs in Qira black sheep and Hetian sheep ovaries using Solexa sequencing. Genet. Mol. Res. 2015, 14, 7356–7367. [Google Scholar] [CrossRef]
- Yang, K.; Shen, J.; Chen, S.W.; Qin, J.; Zheng, X.Y.; Xie, L.P. Upregulation of PAWR by small activating RNAs induces cell apoptosis in human prostate cancer cells. Oncol. Rep. 2016, 35, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, I.H.; Santana, P.; Gonzalez-Robayna, I.; Ferrer, M.; Morales, V.; Blanco, F.L.; Fanjul, L.F. Regulation of the expression of prostate apoptosis response protein 4 (Par-4) in rat granulosa cells. Apoptosis 2007, 12, 769–779. [Google Scholar] [CrossRef]
- Rao, J.U.; Shah, K.B.; Puttaiah, J.; Rudraiah, M. Gene expression profiling of preovulatory follicle in the buffalo cow: Effects of increased IGF-I concentration on periovulatory events. PLoS ONE 2011, 6, e20754. [Google Scholar] [CrossRef] [Green Version]
- Spencer, T.E.; Burghardt, R.C.; Johnson, G.A.; Bazer, F.W. Conceptus signals for establishment and maintenance of pregnancy. Anim. Reprod. Sci. 2004, 82–83, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Verver, D.E.; Langedijk, N.S.; Jordan, P.W.; Repping, S.; Hamer, G. The SMC5/6 complex is involved in crucial processes during human spermatogenesis. Biol. Reprod. 2014, 91, 22. [Google Scholar] [CrossRef] [Green Version]
- Hwang, G.; Verver, D.E.; Handel, M.A.; Hamer, G.; Jordan, P.W. Depletion of SMC5/6 sensitizes male germ cells to DNA damage. Mol. Biol. Cell 2018, 29, 3003–3016. [Google Scholar] [CrossRef]
- Hwang, G.; Sun, F.; O’Brien, M.; Eppig, J.J.; Handel, M.A.; Jordan, P.W. SMC5/6 is required for the formation of segregation-competent bivalent chromosomes during meiosis I in mouse oocytes. Development 2017, 144, 1648–1660. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Li, X.; Sun, H.; Cao, Z.; Gao, R.; Niu, T.; Wang, Y.; Ma, T.; Chen, R.; Wang, C.; et al. Murine Placental-Fetal phosphate dyshomeostasis caused by an xpr1 deficiency accelerates placental calcification and restricts fetal growth in late gestation. J. Bone Miner. Res. 2020, 35, 116–129. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Rodriguez, J.M.; Martin-Cano, F.E.; Ortega-Ferrusola, C.; Masot, J.; Redondo, E.; Gazquez, A.; Gil, M.C.; Aparicio, I.M.; Rojo-Dominguez, P.; Tapia, J.A.; et al. The incorporation of cystine by the soluble carrier family 7 member 11 (SLC7A11) is a component of the redox regulatory mechanism in stallion spermatozoadagger. Biol. Reprod. 2019, 101, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, S.; Mauro, A.; Cellini, V.; Patacchiola, F. The role of Akt signalling in the mammalian ovary. Int. J. Dev. Biol. 2012, 56, 809–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Yan, W.; Shen, W.; Cheng, J.; Xi, Y.; Yuan, S.; Fu, F.; Ding, T.; Luo, A.; Wang, S. Low expression of SEMA6C accelerates the primordial follicle activation in the neonatal mouse ovary. J. Cell. Mol. Med. 2018, 22, 486–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Hama, K.; Contos, J.J.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Aoki, J.; Bandoh, K.; Inoue, A.; Endo, T.; Amano, T.; Suzuki, H.; Arai, H. Lysophosphatidic receptor, LPA3, is positively and negatively regulated by progesterone and estrogen in the mouse uterus. Life Sci. 2006, 79, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Liszewska, E.; Reinaud, P.; Dubois, O.; Charpigny, G. Lysophosphatidic acid receptors in ovine uterus during estrous cycle and early pregnancy and their regulation by progesterone. Domest. Anim. Endocrinol. 2012, 42, 31–42. [Google Scholar] [CrossRef]
- Abedpour, N.; Salehnia, M.; Ghorbanmehr, N. Effect of lysophosphatidic acid on the follicular development and the expression of lysophosphatidic acid receptor genes during in vitro culture of mouse ovary. Vet. Res. Forum. 2018, 9, 59–66. [Google Scholar]
- Buhi, W.C.; Alvarez, I.M.; Kouba, A.J. Oviductal regulation of fertilization and early embryonic development. J. Reprod. Fertil. Suppl. 1997, 52, 285–300. [Google Scholar]
- Mondejar, I.; Acuna, O.S.; Izquierdo-Rico, M.J.; Coy, P.; Aviles, M. The oviduct: Functional genomic and proteomic approach. Reprod. Domest. Anim. 2012, 47 (Suppl. 3), 22–29. [Google Scholar] [CrossRef]
- Sinderewicz, E.; Grycmacher, K.; Boruszewska, D.; Kowalczyk-Zieba, I.; Yamamoto, Y.; Yoshimoto, Y.; Woclawek-Potocka, I. Lysophosphatidic acid synthesis and its receptors’ expression in the bovine oviduct during the oestrous cycle. Reprod. Domest. Anim. 2016, 51, 541–549. [Google Scholar] [CrossRef]
- Da, S.R.; Yang, M.Y.; Caixeta, E.S.; Castilho, A.C.; Amorim, R.L.; Price, C.A.; Fortune, J.E.; Buratini, J. Fibroblast growth factor 18 regulates steroidogenesis in fetal bovine ovarian tissue in vitro. Mol. Reprod. Dev. 2019, 86, 166–174. [Google Scholar]
- Portela, V.M.; Dirandeh, E.; Guerrero-Netro, H.M.; Zamberlam, G.; Barreta, M.H.; Goetten, A.F.; Price, C.A. The role of fibroblast growth factor-18 in follicular atresia in cattle. Biol. Reprod. 2015, 92, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portela, V.M.; Machado, M.; Buratini, J.J.; Zamberlam, G.; Amorim, R.L.; Goncalves, P.; Price, C.A. Expression and function of fibroblast growth factor 18 in the ovarian follicle in cattle. Biol. Reprod. 2010, 83, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, H.J.; Connors, J.M.; Altman, S.N.; Hileman, S.M.; Holaskova, I.; Lehman, M.N.; Mcmanus, C.J.; Nestor, C.C.; Jacobs, B.H.; Goodman, R.L. Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep. Endocrinology 2010, 151, 3836–3846. [Google Scholar] [CrossRef] [Green Version]
- Corander, M.P.; Challis, B.G.; Thompson, E.L.; Jovanovic, Z.; Loraine, T.Y.; Rimmington, D.; Huhtaniemi, I.T.; Murphy, K.G.; Topaloglu, A.K.; Yeo, G.S.; et al. The effects of neurokinin B upon gonadotrophin release in male rodents. J. Neuroendocrinol. 2010, 22, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.D.; Abreu, A.P.; Xu, S.; Muyide, T.; Gianetti, E.; Tusset, C.; Carroll, J.; Latronico, A.C.; Seminara, S.B.; Carroll, R.S.; et al. TACR3 mutations disrupt NK3R function through distinct mechanisms in GnRH-deficient patients. FASEB J. 2014, 28, 1924–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emori, C.; Sugiura, K. Role of oocyte-derived paracrine factors in follicular development. Anim. Sci. J. 2014, 85, 627–633. [Google Scholar] [CrossRef]
- Khalaf, M.; Morera, J.; Bourret, A.; Reznik, Y.; Denoual, C.; Herlicoviez, M.; Mittre, H.; Benhaim, A. BMP system expression in GCs from polycystic ovary syndrome women and the in vitro effects of BMP4, BMP6, and BMP7 on GC steroidogenesis. Eur. J. Endocrinol. 2013, 168, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.M.; Qiao, J.; Leung, P.C. Oocyte-somatic cell interactions in the human ovary-novel role of bone morphogenetic proteins and growth differentiation factors. Hum. Reprod. Update 2016, 23, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochab, A.K.; Extavour, C.G. Bone Morphogenetic Protein (BMP) signaling in animal reproductive system development and function. Dev. Biol. 2017, 427, 258–269. [Google Scholar] [CrossRef]
- Nio-Kobayashi, J.; Trendell, J.; Giakoumelou, S.; Boswell, L.; Nicol, L.; Kudo, M.; Sakuragi, N.; Iwanaga, T.; Duncan, W.C. Bone morphogenetic proteins are mediators of luteolysis in the human corpus luteum. Endocrinology 2015, 156, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Su, Y.Q.; Eppig, J.J. Does bone morphogenetic protein 6 (BMP6) affect female fertility in the mouse? Biol. Reprod. 2010, 83, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Findlay, J.K.; Drummond, A.E.; Dyson, M.L.; Baillie, A.J.; Robertson, D.M.; Ethier, J.F. Recruitment and development of the follicle; The roles of the transforming growth factor-beta superfamily. Mol. Cell. Endocrinol. 2002, 191, 35–43. [Google Scholar] [CrossRef]
- Pangas, S.A.; Li, X.; Robertson, E.J.; Matzuk, M.M. Premature luteinization and cumulus cell defects in ovarian-specific Smad4 knockout mice. Mol. Endocrinol. 2006, 20, 1406–1422. [Google Scholar] [CrossRef] [Green Version]
- Pangas, S.A. Regulation of the ovarian reserve by members of the transforming growth factor beta family. Mol. Reprod. Dev. 2012, 79, 666–679. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.L.; Kaitu’U-Lino, T.J.; Nie, G.; Sanchez-Partida, L.G.; Findlay, J.K.; Salamonsen, L.A. Complex expression patterns support potential roles for maternally derived activins in the establishment of pregnancy in mouse. Reproduction 2006, 132, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Vassalli, A.; Matzuk, M.M.; Gardner, H.A.; Lee, K.F.; Jaenisch, R. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994, 8, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochstrasser, M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 1995, 7, 215–223. [Google Scholar] [CrossRef]
- Connelly, C.; Hieter, P. Budding yeast SKP1 encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell 1996, 86, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Yang, M.; Gao, C.; Yue, W.; Liang, X.; Xie, B.; Zhu, X.; Fan, S.; Li, R.; Li, M. Fbxo30 regulates chromosome segregation of oocyte meiosis. Cell. Mol. Life Sci. 2019, 76, 2217–2229. [Google Scholar] [CrossRef]
- Kinterova, V.; Kanka, J.; Petruskova, V.; Toralova, T. Inhibition of Skp1-Cullin-F-box complexes during bovine oocyte maturation and preimplantation development leads to delayed development of embryosdagger. Biol. Reprod. 2019, 100, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Kim, T.H.; Choi, H.; Bae, C.H.; Yang, Y.M.; Baek, J.A.; Lee, J.C.; Cho, E.S. Disruption of Tgfbr2 in odontoblasts leads to aberrant pulp calcification. J. Dent. Res. 2015, 94, 828–835. [Google Scholar] [CrossRef]
- Wang, W.; La, Y.; Zhou, X.; Zhang, X.; Li, F.; Liu, B. The genetic polymorphisms of TGFbeta superfamily genes are associated with litter size in a Chinese indigenous sheep breed (Hu sheep). Anim. Reprod. Sci. 2018, 189, 19–29. [Google Scholar] [CrossRef]
- Song, C.; Yang, J.; Jiang, R.; Yang, Z.; Li, H.; Huang, Y.; Lan, X.; Lei, C.; Ma, Y.; Qi, X.; et al. MiR-148a-3p regulates proliferation and apoptosis of bovine muscle cells by targeting KLF6. J. Cell. Physiol. 2019, 234, 15742–15750. [Google Scholar] [CrossRef]
- Manaster, I.; Goldman-Wohl, D.; Greenfield, C.; Nachmani, D.; Tsukerman, P.; Hamani, Y.; Yagel, S.; Mandelboim, O. MiRNA-mediated control of HLA-G expression and function. PLoS ONE 2012, 7, e33395. [Google Scholar] [CrossRef]
- Frazier, S.; Mcbride, M.W.; Mulvana, H.; Graham, D. From animal models to patients: The role of placental microRNAs, miR-210, miR-126, and miR-148a/152 in preeclampsia. Clin. Sci. 2020, 134, 1001–1025. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, M.; Gao, R.; Xie, S.; Sun, X.; He, J.; Chen, X.; Li, Q.; Lu, S.; Yang, M.; et al. Identification of differentially expressed miRNAs in serum extracellular vesicles (EVs) of Kazakh sheep at early pregnancy. Reprod. Domest. Anim. 2021, 56, 713–724. [Google Scholar] [CrossRef]
- Ito, M.; Sferruzzi-Perri, A.N.; Edwards, C.A.; Adalsteinsson, B.T.; Allen, S.E.; Loo, T.H.; Kitazawa, M.; Kaneko-Ishino, T.; Ishino, F.; Stewart, C.L.; et al. A trans-homologue interaction between reciprocally imprinted miR-127 and Rtl1 regulates placenta development. Development 2015, 142, 2425–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, C.; Wang, S.; Yin, C.; Liu, L.; Zhou, L.; Ma, Y. Loss of hsa_circ_0118530 inhibits human granulosa-like tumor cell line KGN cell injury by sponging miR-136. Gene 2020, 744, 144591. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, Y.; Nakamura, K.; Kogure, K.; Minegishi, T. Role of microRNA-136-3p on the expression of luteinizing hormone-human chorionic gonadotropin receptor mRNA in rat ovaries. Biol. Reprod. 2013, 89, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Cheng, Y.; Zhang, Y.; Liao, S.; Yin, T.; Yang, J. The miR-27a-3p/USP25 axis participates in the pathogenesis of recurrent miscarriage by inhibiting trophoblast migration and invasion. J. Cell. Physiol. 2019, 234, 19951–19963. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xu, B.; Wu, J. Circular RNA expression profiles of mouse ovaries during postnatal development and the function of circular RNA epidermal growth factor receptor in granulosa cells. Metabolism 2018, 85, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Chen, X.; Liu, M.; Zhang, L.; Ma, X.; Tian, S. Differential expression and functional analysis of CircRNA in the ovaries of low and high fecundity hanper sheep. Animals 2021, 11, 1863. [Google Scholar] [CrossRef]
- Ernst, J.; Grabiec, U.; Greither, T.; Fischer, B.; Dehghani, F. The endocannabinoid system in the human granulosa cell line KGN. Mol. Cell. Endocrinol. 2016, 423, 67–76. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Gao, B.W.; Guo, H.X.; Ren, Q.L.; Wang, X.W.; Chen, J.F.; Wang, J.; Zhang, Z.J.; Ma, Q.; Xing, B.S. MiR-181a promotes porcine granulosa cell apoptosis by targeting TGFBR1 via the activin signaling pathway. Mol. Cell. Endocrinol. 2020, 499, 110603. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′–3′) | Product Size (bp) |
|---|---|---|
| novel_circ_0000417 | F: GAGCAAACTCGCTTCTGCATT | 226 |
| R: CTCTCTCTGCGGTTTTGTGGT | ||
| novel_circ_0000962 | F: TGTCAAGCAGTGGGTTAATCAGA | 152 |
| R: CATTAACGGCTGGTCTGGGT | ||
| novel_circ_0005109 | F: AGAACTGAGAAAAAGGTCAGACT | 111 |
| R: CATACATTCTGGCGCTCGTG | ||
| novel_circ_0016586 | F: ATCAGAGAAGCCACCAGGAAC | 128 |
| R: GCTTCTTACAGAGCTGGGGTC | ||
| novel_circ_0017815 | F: AAACGATGAAGATGCTGAGCC | 134 |
| R: GCTCCCGAGGTTTGATGTCC | ||
| β-Actin | F: CCAACCGTGAGAAGATGACC | 97 |
| R: CCCGAGGCGTACAGGGACAG | ||
| oar-miR-10b | F: GCGCACCCTGTAGAACCGAATTTGTG | 22 |
| oar-miR-376b-3p | F: GCGCGCATCATAGAGGAAAATCCATGT | 21 |
| oar-miR-376c-3p | F: GCGCGAACATAGAGGAAATTCCACGT | 21 |
| oar-miR-543-3p | F: GCGAAACATTCGCGGTGCACTTCTTT | 23 |
| oar-miR-665-3p | F: ACCAGTAGGCCGAGGCCC | 22 |
| oar-miR-U6 | F: CAAGGATGACACGCAAATTCG | 21 |
| Sample Name | Raw Reads | Clean Reads | Total Mapped | Q30 (%) | Aligned Rate (%) | GC Content (%) |
|---|---|---|---|---|---|---|
| MM_F_O_1 | 96,196,362 | 94,744,878 | 87,141,547 | 94.86 | 91.97 | 47.56 |
| MM_F_O_2 | 92,640,970 | 90,020,528 | 82,821,574 | 93.89 | 92 | 48.67 |
| MM_F_O_3 | 93,009,358 | 90,394,288 | 80,814,953 | 93.88 | 89.4 | 50.38 |
| MM_L_O_1 | 88,588,260 | 85,274,304 | 74,294,998 | 92.5 | 87.12 | 47.62 |
| MM_L_O_2 | 97,001,262 | 93,030,528 | 83,370,890 | 92.82 | 89.62 | 48.06 |
| MM_L_O_3 | 96,027,084 | 92,170,182 | 78,243,273 | 92.77 | 84.89 | 50.11 |
| ww_F_O_1 | 90,181,468 | 87,668,706 | 79,148,368 | 94.1 | 90.28 | 49.15 |
| ww_F_O_2 | 109,389,164 | 106,239,400 | 94,022,401 | 94.19 | 88.5 | 50.44 |
| ww_F_O_3 | 84,735,812 | 81,446,054 | 73,026,516 | 93.15 | 89.66 | 48.51 |
| ww_L_O_1 | 102,279,454 | 100,207,792 | 92,162,400 | 93.47 | 91.97 | 49.53 |
| ww_L_O_2 | 94,330,442 | 90,209,692 | 81,346,141 | 93.23 | 90.17 | 48.46 |
| ww_L_O_3 | 102,550,798 | 99,314,822 | 89,937,364 | 93.03 | 90.56 | 48.39 |
| Sample Name | Raw Reads | Clean Reads | Screened Reads | Total Mapped | Q30 (%) | Aligned Rate (%) |
|---|---|---|---|---|---|---|
| MM_F_O_1 | 14,546,606 | 14,321,558 | 13,569,930 | 12,570,515 | 96.53 | 92.64 |
| MM_F_O_2 | 13,014,433 | 12,763,844 | 11,630,660 | 10,792,527 | 96.42 | 92.79 |
| MM_F_O_3 | 12,371,162 | 12,148,054 | 11,091,183 | 9,918,242 | 94.81 | 89.42 |
| MM_L_O_1 | 12,625,788 | 12,431,487 | 11,385,564 | 10,489,170 | 94.23 | 92.13 |
| MM_L_O_2 | 11,973,021 | 11,819,020 | 11,119,789 | 10,130,283 | 94.02 | 91.10 |
| MM_L_O_3 | 10,296,511 | 10,176,943 | 9,946,122 | 8,720,780 | 95.77 | 87.68 |
| ww_F_O_1 | 12,163,978 | 11,962,622 | 10,823,047 | 9,705,421 | 94.68 | 89.67 |
| ww_F_O_2 | 11,447,533 | 11,252,977 | 10,467,663 | 9,283,625 | 94.84 | 88.69 |
| ww_F_O_3 | 13,365,195 | 13,090,842 | 11,603,455 | 10,748,779 | 93.56 | 92.63 |
| ww_L_O_1 | 12,185,470 | 11,826,433 | 11,381,948 | 10,275,416 | 95.50 | 90.28 |
| ww_L_O_2 | 11,800,083 | 11,594,302 | 11,237,422 | 10,177,899 | 95.70 | 90.57 |
| ww_L_O_3 | 11,410,997 | 11,179,464 | 10,446,590 | 9,313,721 | 95.39 | 89.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; He, X.; Zhang, X.; Zhang, J.; Guo, X.; Sun, W.; Chu, M. Analysis of Expression Profiles of CircRNA and MiRNA in Oviduct during the Follicular and Luteal Phases of Sheep with Two Fecundity (FecB Gene) Genotypes. Animals 2021, 11, 2826. https://doi.org/10.3390/ani11102826
Li Z, He X, Zhang X, Zhang J, Guo X, Sun W, Chu M. Analysis of Expression Profiles of CircRNA and MiRNA in Oviduct during the Follicular and Luteal Phases of Sheep with Two Fecundity (FecB Gene) Genotypes. Animals. 2021; 11(10):2826. https://doi.org/10.3390/ani11102826
Chicago/Turabian StyleLi, Zhifeng, Xiaoyun He, Xiaosheng Zhang, Jinlong Zhang, Xiaofei Guo, Wei Sun, and Mingxing Chu. 2021. "Analysis of Expression Profiles of CircRNA and MiRNA in Oviduct during the Follicular and Luteal Phases of Sheep with Two Fecundity (FecB Gene) Genotypes" Animals 11, no. 10: 2826. https://doi.org/10.3390/ani11102826
APA StyleLi, Z., He, X., Zhang, X., Zhang, J., Guo, X., Sun, W., & Chu, M. (2021). Analysis of Expression Profiles of CircRNA and MiRNA in Oviduct during the Follicular and Luteal Phases of Sheep with Two Fecundity (FecB Gene) Genotypes. Animals, 11(10), 2826. https://doi.org/10.3390/ani11102826