Genomic Analysis Using Bayesian Methods under Different Genotyping Platforms in Korean Duroc Pigs

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genotype and Phenotype Data Editing and Imputation
2.2. Deregression of Expected Breeding Values (DEBVs) for Response Variables
2.3. Statistical Method for Estimating SNP Effects
2.4. Identification of Significant Window Regions and SNP Markers
2.5. Accuracy of Genomic Prediction under a 10-Fold Cross-Validation
3. Results and Discussion
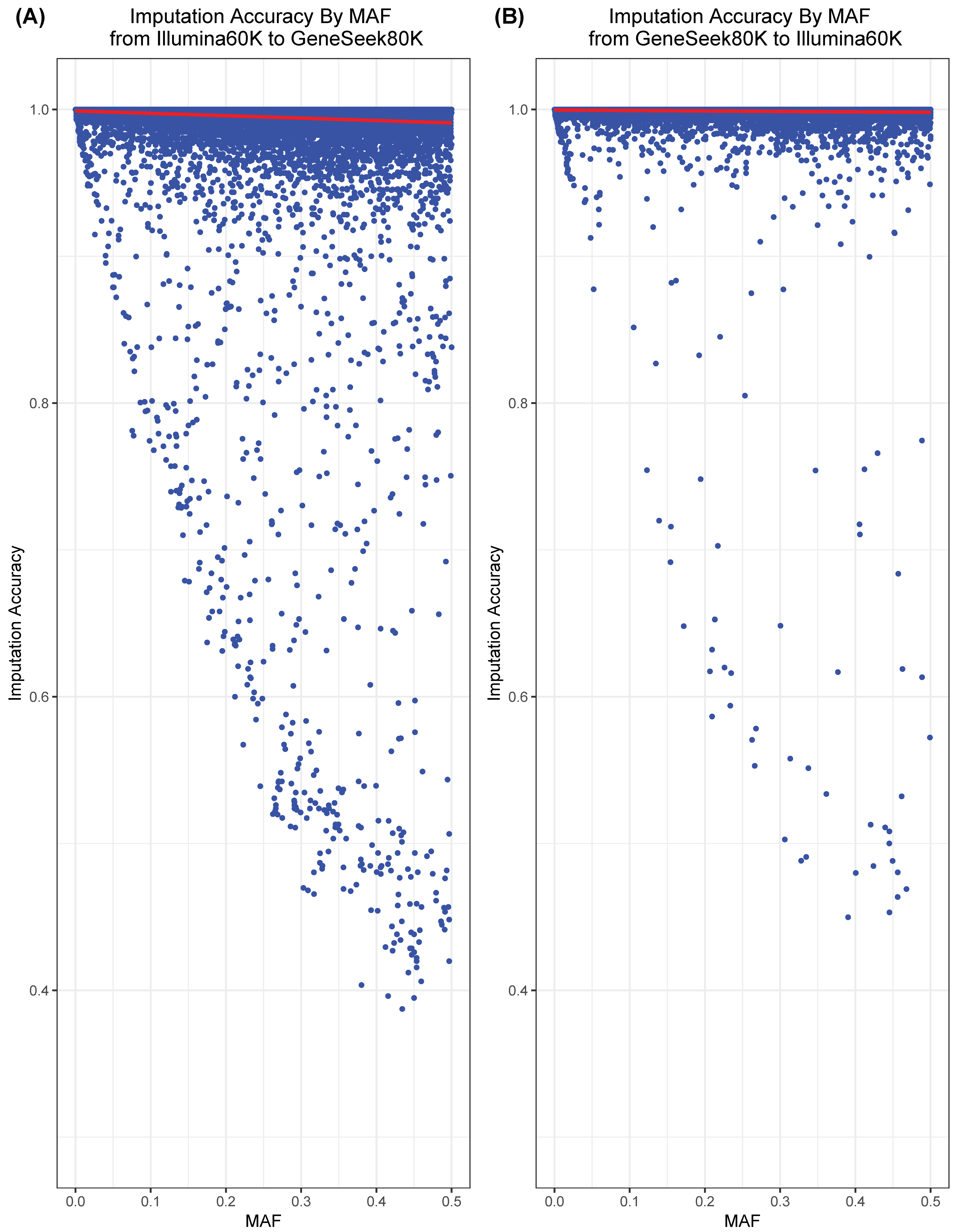
3.1. Assessing the Accuracy of Imputation
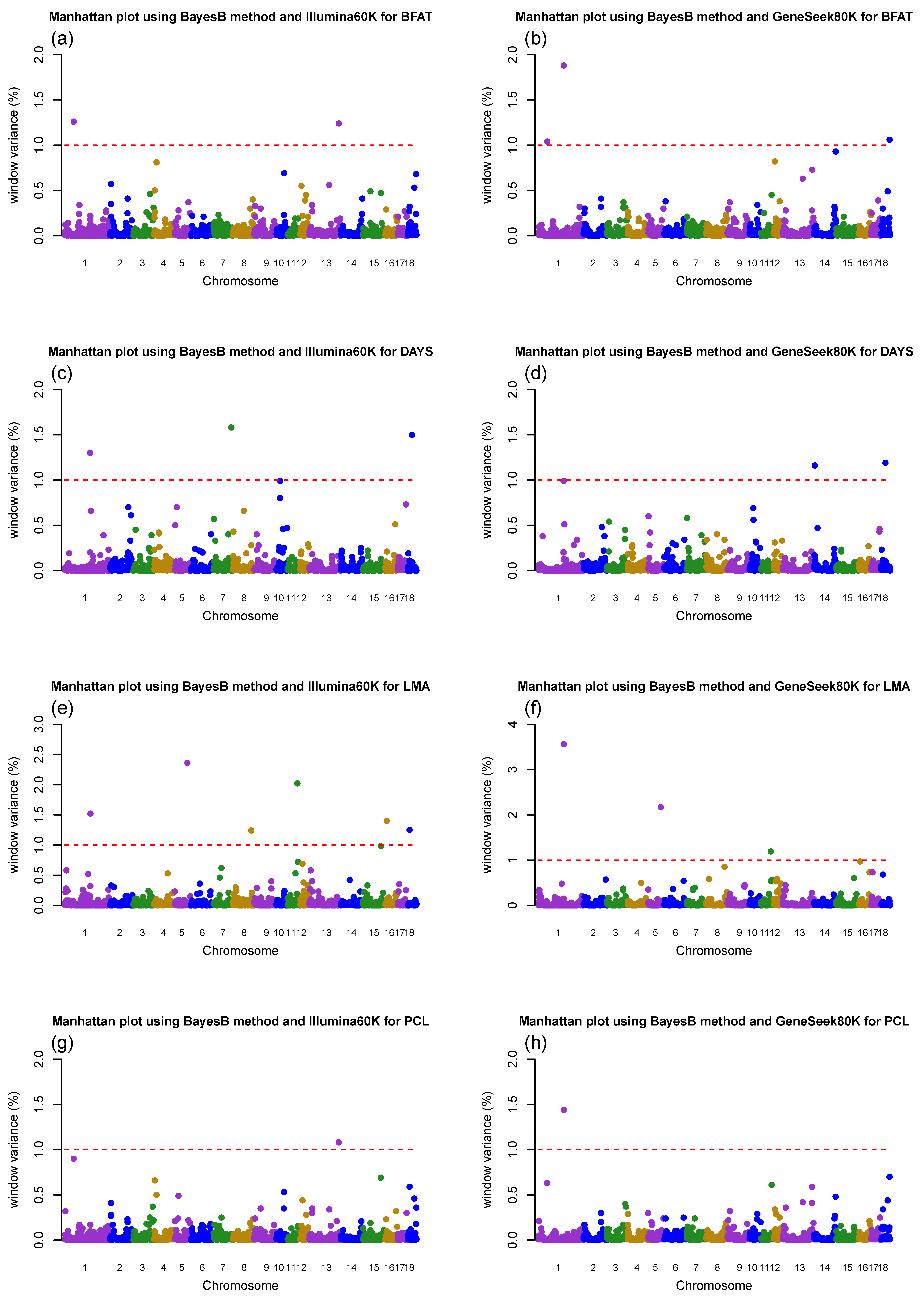
3.2. Genome-Wide Association Study (GWAS) for Growth- and Production-Related Traits
3.3. Accuracy of Genomic Prediction
3.3.1. SNP Genotyping Platforms and Bayesian Methods
3.3.2. Response Variables (DEBVincPA and DEBVexcPA)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kachman, S.D. Incorporation of marker scores into national genetic evaluations. In Proceedings of the 9th Genetic Prediction Workshop, Beef Improvement Federation, Kansas City, MO, USA, 8–10 December 2008; pp. 92–98. [Google Scholar]
- Samore, A.B.; Fontanesi, L. Genomic selection in pigs: State of the art and perspectives. Ital. J. Anim. Sci. 2016, 15, 211–232. [Google Scholar] [CrossRef]
- Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genet 2001, 157, 1819–1829. [Google Scholar]
- Van Raden, P.M.; Sullivan, P.G. International genomic evaluation methods for dairy cattle. Genet. Sel. Evol. 2010, 42, 7. [Google Scholar] [CrossRef] [PubMed]
- Erbe, M.; Hayes, B.; Matukumalli, L.; Goswami, S.; Bowman, P.J.; Reich, C.M.; Mason, B.A.; Goddard, M.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef]
- Su, G.; Brøndum, R.F.; Ma, P.; Gulbrandtsen, B.; Aamand, G.P.; Lund, M.S. Comparison of genomic predictions using medium-density (~54,000) and high-density (~777,000) single nucleotide polymorphism marker panels in Nordic Holstein and Red Dairy Cattle populations. J. Dairy Sci. 2012, 95, 4657–46465. [Google Scholar] [CrossRef]
- Pérez-Enciso, M.; Rincón, J.C.; Legarra, A. Sequence-vs. chip-assisted genomic selection: Accurate biological information is advised. Genet. Sel. Evol. 2015, 47, 43. [Google Scholar] [CrossRef]
- Lee, J.; Kachman, S.D.; Spangler, M.L. The impact of training strategies on the accuracy of genomic predictors in United States Red Angus cattle. J. Anim. Sci. 2017, 95, 3406–3414. [Google Scholar] [CrossRef]
- Saatchi, M.; McClure, M.C.; McKay, S.D.; Rolf, M.M.; Kim, J.; Decker, J.E.; Taxis, T.M.; Chapple, R.H.; Ramey, H.R.; Northcutt, S.L.; et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet. Sel. Evol. 2011, 43, 40. [Google Scholar] [CrossRef]
- Hess, M.; Druet, T.; Hess, A.; Garrick, D. Fixed-length haplotypes can improve genomic prediction accuracy in an admixed dairy cattle population. Genet. Sel. Evol. 2017, 49, 54. [Google Scholar] [CrossRef]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Van den Berg, I.; Boichard, D.; Lund, M.S. Sequence variants selected from a multi-breed GWAS can improve the reliability of genomic predictions in dairy cattle. Genet. Sel. Evol. 2016, 48, 83. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei, M.; Chesnais, J.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, A.; Gogel, B.; Cullis, B.; Welham, S.; Thompson, R. ASReml User Guide Release 4.1 Structural Specification; VSN International Ltd.: Hemel Hempstead, UK, 2015. [Google Scholar]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; Schnabel, R.D.; Rolf, M.M.; Taylor, J.F.; Garrick, D.J. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet. Sel. Evol. 2012, 44, 38. [Google Scholar] [CrossRef]
- Kizilkaya, K.; Fernando, R.; Garrick, D. Genomic prediction of simulated multibreed and purebred performance using observed fifty thousand single nucleotide polymorphism genotypes. J. Anim. Sci. 2010, 88, 544–551. [Google Scholar] [CrossRef]
- Garrick, D.J.; Fernando, R.L. Implementing a QTL detection study (GWAS) using genomic prediction methodology. In Genome-Wide Association Studies and Genomic Prediction; Gondro, C., van der Werf, J., Hayes, B., Eds.; Springer Science + Business Media, LLC: Totowa, NJ, USA, 2013; pp. 275–298. [Google Scholar]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Badke, Y.M.; Bates, R.O.; Ernst, C.W.; Schwab, C.; Fix, J.; Van Tassell, C.P.; Steibel, J.P. Methods of tagSNP selection and other variables affecting imputation accuracy in swine. BMC Genet. 2013, 14, 8. [Google Scholar] [CrossRef]
- Ma, P.; Brøndum, R.F.; Zhang, Q.; Lund, M.S.; Su, G. Comparison of different methods for imputing genome-wide marker genotypes in Swedish and Finnish Red Cattle. J. Dairy Sci. 2013, 96, 4666–4677. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009, 84, 210–223. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M.; O’Connell, J.R.; Wiggans, G.R.; Weigel, K.A. Genomic evaluations with many more genotypes. Genet. Sel. Evol. 2011, 43, 10. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.M.; Kinghorn, B.P.; Tier, B.; van der Werf, J.H.; Cleveland, M.A. A phasing and imputation method for pedigreed populations that results in a single-stage genomic evaluation. Genet. Sel. Evol. 2012, 44, 9. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, C.; Srinivasan, S.; Shapiro, A.; Zhang, S.; Picard, F.; Clement, K.; Lubrano-Berthelier, C.; Vaisse, C. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides 2005, 26, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Adan, R.A.; Tiesjema, B.; Hillebrand, J.J.; la Fleur, S.E.; Kas, M.J.; de Krom, M. The MC4 receptor and control of appetite. Br. J. Pharmacol. 2006, 149, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Onteru, S.; Plastow, G.; Rothschild, M. Detailed characterization of the porcine MC4R gene in relation to fatness and growth. Anim. Genet. 2009, 40, 401–409. [Google Scholar] [CrossRef]
- Kim, K.S.; Larsen, N.; Short, T.; Plastow, G.; Rothschild, M.F. A missense variant of the porcine melanocortin-4 receptor (MC4R) gene is associated with fatness, growth, and feed intake traits. Mamm. Genome 2000, 11, 131–135. [Google Scholar] [CrossRef]
- Barb, C.; Robertson, A.; Barrett, J.; Kraeling, R.; Houseknecht, K. The role of melanocortin-3 and-4 receptor in regulating appetite, energy homeostasis and neuroendocrine function in the pig. J. Endocrinol. 2004, 181, 39–52. [Google Scholar] [CrossRef]
- Kim, K.-S.; Reecy, J.; Hsu, W.; Anderson, L.; Rothschild, M. Functional and phylogenetic analyses of a melanocortin-4 receptor mutation in domestic pigs. Domest. Anim. Endocrinol. 2004, 26, 75–86. [Google Scholar] [CrossRef]
- Guo, G.; Lund, M.S.; Zhang, Y.; Su, G. Comparison between genomic predictions using daughter yield deviation and conventional estimated breeding value as response variables. J. Anim. Breed. Genet. 2010, 127, 423–432. [Google Scholar] [CrossRef]
- Boddhireddy, P.; Kelly, M.; Northcutt, S.; Prayaga, K.C.; Rumph, J.; DeNise, S. Genomic predictions in Angus cattle: Comparisons of sample size, response variables, and clustering methods for cross-validation. J. Anim. Sci. 2014, 92, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, H.; Fernando, R.L.; Garrick, D.J.; Taylor, J. Characterization of the F94L double muscling mutation in pure-and crossbred Limousin animals. Anim. Ind. Rep. 2015, 661, 19. [Google Scholar]
- Lee, J.; Lee, S.; Park, J.-E.; Moon, S.-H.; Choi, S.-W.; Go, G.-W.; Lim, D.; Kim, J.-M. Genome-wide association study and genomic predictions for exterior traits in Yorkshire pigs. J. Anim. Sci. 2019, 97, 2793–2802. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trait 1 | Additive Genetic Variance | Phenotypic Variance | Heritability |
|---|---|---|---|
| BFAT | 1.21 | 3.42 | 0.35 |
| DAYS | 34.57 | 85.10 | 0.41 |
| LMA | 1.14 | 7.15 | 0.16 |
| PCL | 2.09 | 5.52 | 0.38 |
| No. of Clusters | No. of Animals | inBreC 1 | amax_within2 | amax_between3 | aij_within4 | aij_between5 |
|---|---|---|---|---|---|---|
| 1 | 94 | 0.011 | 0.48 (0.11) | 0.32 (0.14) | 0.09 (0.02) | 0.05 (0.01) |
| 2 | 162 | 0.031 | 0.47 (0.12) | 0.17 (0.10) | 0.07 (0.01) | 0.01 (0.01) |
| 3 | 78 | 0.048 | 0.52 (0.10) | 0.37 (0.13) | 0.19 (0.03) | 0.05 (0.00) |
| 4 | 113 | 0.070 | 0.54 (0.11) | 0.40 (0.11) | 0.19 (0.03) | 0.05 (0.00) |
| 5 | 61 | 0.048 | 0.52 (0.11) | 0.39 (0.14) | 0.17 (0.03) | 0.05 (0.01) |
| 6 | 65 | 0.053 | 0.55 (0.08) | 0.37 (0.13) | 0.23 (0.02) | 0.05 (0.01) |
| 7 | 70 | 0.029 | 0.42 (0.15) | 0.39 (0.12) | 0.10 (0.03) | 0.04 (0.01) |
| 8 | 112 | 0.009 | 0.49 (0.10) | 0.26 (0.10) | 0.10 (0.03) | 0.03 (0.00) |
| 9 | 123 | 0.063 | 0.54 (0.10) | 0.19 (0.08) | 0.17 (0.02) | 0.03 (0.01) |
| 10 | 94 | 0.001 | 0.34 (0.19) | 0.12 (0.14) | 0.03 (0.02) | 0.01 (0.01) |
| Trait 1 | SSC _Mb | GV (%) 2 | Informative SNP | Position (Mb) | Effect | BF 3 | Region Annotation | Gene Annotation |
|---|---|---|---|---|---|---|---|---|
| BFAT | 1_62 | 1.26 | MARC0038944 | 62.12 | −0.04 | 24.17 | intergenic | CGA (dist = 131054) |
| ALGA0003581 | 62.20 | −0.04 | 23.39 | intergenic | CGA (dist = 44656) | |||
| ALGA0003583 | 62.23 | −0.04 | 22.52 | intergenic | CGA (dist = 16224) | |||
| ALGA0003587 | 62.24 | 0.03 | 21.88 | intergenic | CGA (dist = 2593) | |||
| 13_205 | 1.24 | ASGA0059825 | 205.31 | 0.12 | 136.38 | intergenic | CLDN8 (dist = 1144428), SOD1 (dist = 309936) | |
| 4_16 | 0.81 | ASGA0018674 | 16.88 | 0.10 | 97.55 | intergenic | FBXO32 (dist = 210423), DERL1 (dist = 213410) | |
| DAYS | 7_124 | 1.58 | ASGA0093614 | 124.68 | 0.94 | 708.50 | intergenic | BDKRB2 (dist = 26181) |
| 18_29 | 1.50 | ALGA0097693 | 29.01 | 0.97 | 240.97 | intergenic | TSPAN12 (dist = 1290900), CFTR (dist = 1388059) | |
| 1_177 | 1.30 | ASGA0004988 | 177.53 | −0.66 | 85.63 | intergenic | RNF152 (dist = 468819), MC4R (dist = 1019391) | |
| 10_27 | 0.99 | H3GA0029615 | 27.03 | −0.62 | 77.98 | intergenic | MIR181A-1 (dist = 601150), NR5A2 (dist = 252919) | |
| 10_26 | 0.80 | H3GA0029613 | 26.91 | −0.58 | 69.77 | intergenic | MIR181A-1 (dist = 489646), NR5A2 (dist = 364423) | |
| LMA | 5_87 | 2.36 | ALGA0033240 | 87.39 | 0.21 | 934.40 | intergenic | SLC5A8 (dist = 318494), NR1H4 (dist = 399478) |
| 11_68 | 2.02 | CASI0007856 | 68.91 | −0.18 | 864.04 | intergenic | DCT (dist = 757772) | |
| 1_179 | 1.52 | ALGA0006660 | 179.02 | 0.14 | 57.55 | intergenic | PMAIP1 (dist = 161261), MIR122 (dist = 897655) | |
| ALGA0006655 | 179.00 | 0.12 | 47.49 | intergenic | PMAIP1 (dist = 144947), MIR122 (dist = 913969) | |||
| 16_9 | 1.40 | ALGA0101487 | 99.10 | 0.09 | 96.92 | - | NONE | |
| 18_12 | 1.25 | ASGA0078904 | 12.62 | −0.05 | 42.71 | intergenic | ZC3HAV1 (dist = 1552776), PTN (dist = 273756) | |
| M1GA0023069 | 12.64 | 0.05 | 38.94 | intergenic | ZC3HAV1 (dist = 1572284), PTN (dist = 254248) | |||
| 8_128 | 1.24 | ALGA0115575 | 128.24 | −0.13 | 149.93 | intergenic | NFKB1 (dist = 573086), PPP3CA (dist = 224770) | |
| PCL | 13_205 | 1.08 | ASGA0059825 | 205.31 | −0.18 | 190.47 | intergenic | CLDN8 (dist = 1144428), SOD1 (dist = 309936) |
| 1_62 | 0.90 | ALGA0003581 | 62.20 | 0.04 | 21.72 | intergenic | CGA (dist = 44656) | |
| MARC0038944 | 62.12 | 0.04 | 21.00 | intergenic | CGA (dist = 131054) | |||
| ALGA003583 | 62.23 | 0.04 | 20.59 | intergenic | CGA (dist = 16224) |
| Trait 1 | SSC _Mb | GV (%) 2 | Informative SNP | Position (Mb) | Effect | BF 3 | Region Annotation | Gene Annotation |
|---|---|---|---|---|---|---|---|---|
| BFAT | 1_178 | 1.88 | WU_10.2_1_178188861 | 178.19 | −0.21 | 195.56 | intergenic | RNF152(dist = 1123583), MC4R (dist = 364627) |
| 18_58 | 1.06 | WU_10.2_18_58809866 | 58.81 | −0.04 | 26.71 | intergenic | INHBA (dist = 800771) | |
| 1_62 | 1.04 | ALGA0003581 | 62.20 | −0.03 | 21.94 | intergenic | CGA (dist = 44656) | |
| ALGA0003587 | 62.24 | 0.03 | 21.51 | intergenic | CGA (dist = 2593) | |||
| 14_150 | 0.93 | WU_10.2_14_150298075 | 150.30 | 0.08 | 68.20 | intergenic | GLRX3 (dist = 891194) | |
| M1GA0019859 | 150.87 | 0.03 | 21.50 | intergenic | GLRX3 (dist = 891194) | |||
| DAYS | 18_29 | 1.19 | ALGA0097693 | 29.01 | 0.78 | 145.57 | intergenic | TSPAN12 (dist = 1290900), CFTR (dist = 1388059) |
| 14_4 | 1.16 | WU_10.2_14_4968099 | 4.97 | 0.86 | 186.71 | intergenic | LPL (dist = 511359), DOK2 (dist = 1649547) | |
| 1_177 | 0.99 | ASGA0004988 | 177.53 | −0.51 | 58.27 | intergenic | RNF152 (dist = 468819), MC4R (dist = 1019391) | |
| LMA | 1_178 | 3.56 | WU_10.2_1_178188861 | 178.19 | 0.30 | 1010.87 | intergenic | RNF152 (dist = 1123583), MC4R (dist = 364627) |
| 5_87 | 2.17 | ALGA0033240 | 87.39 | 0.20 | 659.04 | intergenic | SLC5A8 (dist = 318494), NR1H4 (dist = 399478) | |
| 11_68 | 1.19 | CASI0007856 | 68.91 | −0.12 | 158.68 | intergenic | DCT (dist = 757772) | |
| 16_9 | 0.97 | ALGA0101487 | 9.91 | 0.08 | 88.43 | - | NONE | |
| 8_128 | 0.85 | ALGA0115575 | 128.24 | −0.09 | 77.91 | intergenic | NFKB1 (dist = 573086), PPP3CA (dist = 224770) | |
| PCL | 1_178 | 1.44 | WU_10.2_1_178188861 | 178.19 | 0.26 | 167.70 | intergenic | RNF152 (dist = 1123583), MC4R (dist = 364627) |
| 11_74 | 0.61 | WU_10.2_11_74507674 | 74.51 | 0.12 | 85.22 | intergenic | IPO5 (dist = 334379), SLC15A1 (dist = 382105) |
| SNP Platforms | Bayes Types | Traits 1 | Response Variables 2 | |
|---|---|---|---|---|
| DEBVexcPA | DEBVincPA | |||
| Illumina60K | BayesB | BFAT | 0.18 (0.044) | 0.25 (0.043) |
| DAYS | 0.19 (0.046) | 0.27 (0.044) | ||
| LMA | 0.23 (0.041) | 0.30 (0.040) | ||
| PCL | 0.22 (0.045) | 0.29 (0.043) | ||
| BayesC | BFAT | 0.18 (0.044) | 0.26 (0.042) | |
| DAYS | 0.19 (0.046) | 0.28 (0.044) | ||
| LMA | 0.23 (0.041) | 0.31 (0.040) | ||
| PCL | 0.22 (0.045) | 0.30 (0.043) | ||
| GeneSeek80K | BayesB | BFAT | 0.18 (0.044) | 0.25 (0.042) |
| DAYS | 0.21 (0.046) | 0.27 (0.044) | ||
| LMA | 0.25 (0.040) | 0.33 (0.040) | ||
| PCL | 0.22 (0.045) | 0.30 (0.043) | ||
| BayesC | BFAT | 0.18 (0.044) | 0.25 (0.042) | |
| DAYS | 0.20 (0.046) | 0.27 (0.044) | ||
| LMA | 0.24 (0.041) | 0.32 (0.040) | ||
| PCL | 0.22 (0.045) | 0.30 (0.043) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kim, Y.; Cho, E.; Cho, K.; Sa, S.; Kim, Y.; Choi, J.; Kim, J.; Hong, J.; Choi, T. Genomic Analysis Using Bayesian Methods under Different Genotyping Platforms in Korean Duroc Pigs. Animals 2020, 10, 752. https://doi.org/10.3390/ani10050752
Lee J, Kim Y, Cho E, Cho K, Sa S, Kim Y, Choi J, Kim J, Hong J, Choi T. Genomic Analysis Using Bayesian Methods under Different Genotyping Platforms in Korean Duroc Pigs. Animals. 2020; 10(5):752. https://doi.org/10.3390/ani10050752
Chicago/Turabian StyleLee, Jungjae, Yongmin Kim, Eunseok Cho, Kyuho Cho, Soojin Sa, Youngsin Kim, Jungwoo Choi, Jinsoo Kim, Junki Hong, and Taejeong Choi. 2020. "Genomic Analysis Using Bayesian Methods under Different Genotyping Platforms in Korean Duroc Pigs" Animals 10, no. 5: 752. https://doi.org/10.3390/ani10050752
APA StyleLee, J., Kim, Y., Cho, E., Cho, K., Sa, S., Kim, Y., Choi, J., Kim, J., Hong, J., & Choi, T. (2020). Genomic Analysis Using Bayesian Methods under Different Genotyping Platforms in Korean Duroc Pigs. Animals, 10(5), 752. https://doi.org/10.3390/ani10050752