Using High-Density SNP Array to Reveal Selection Signatures Related to Prolificacy in Chinese and Kazakhstan Sheep Breeds

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
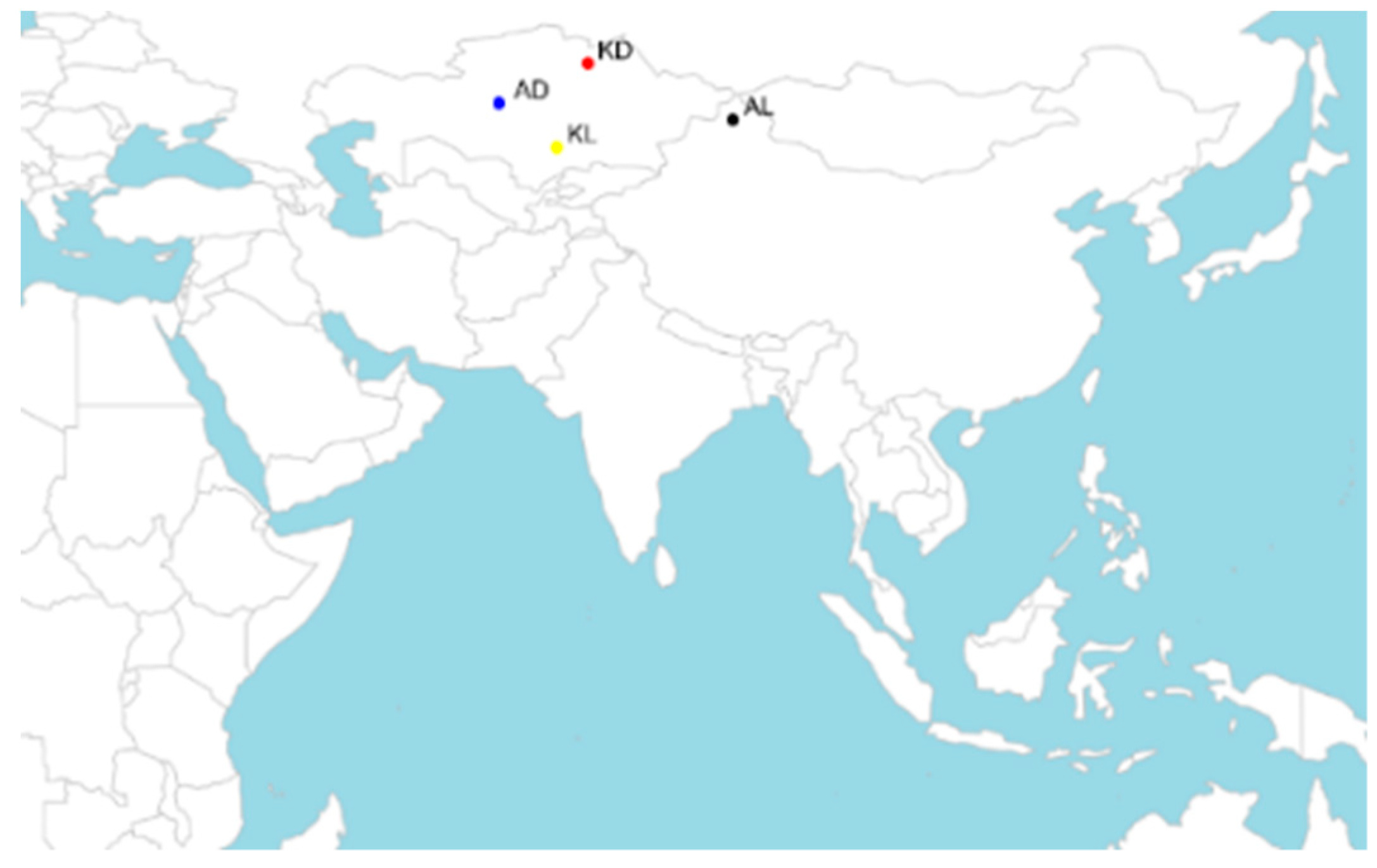
2.1. Samples and Phenotype
2.2. SNP Genotyping and Quality Control
2.3. Principal Components Analysis (PCA), Population Admixture Analysis, and LD Decay
2.4. Identification of Selection Signatures
2.5. Functional Annotation for Selection Signatures
3. Results
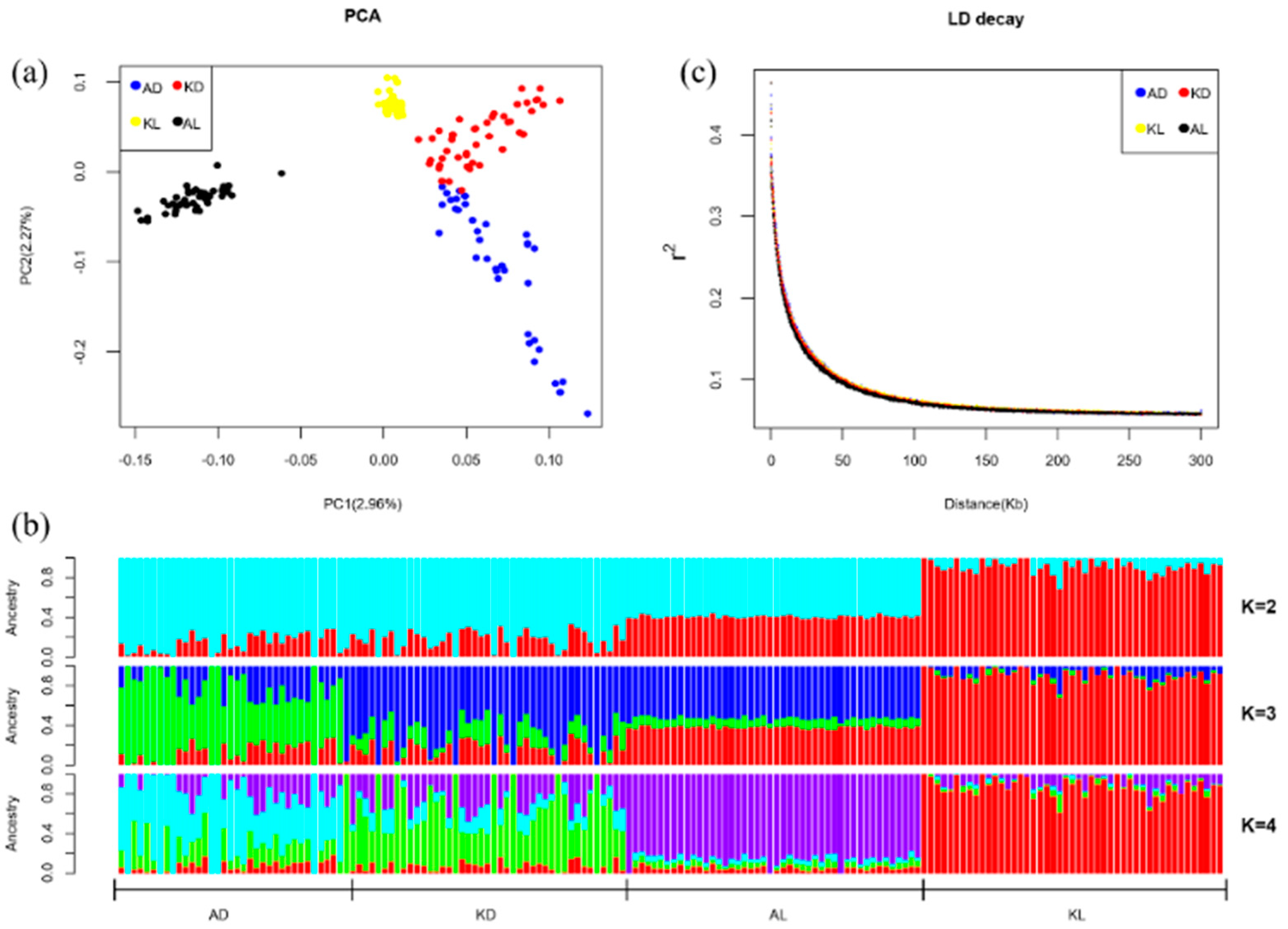
3.1. PCA Analysis, STRUCTURE Analysis and LD Decay
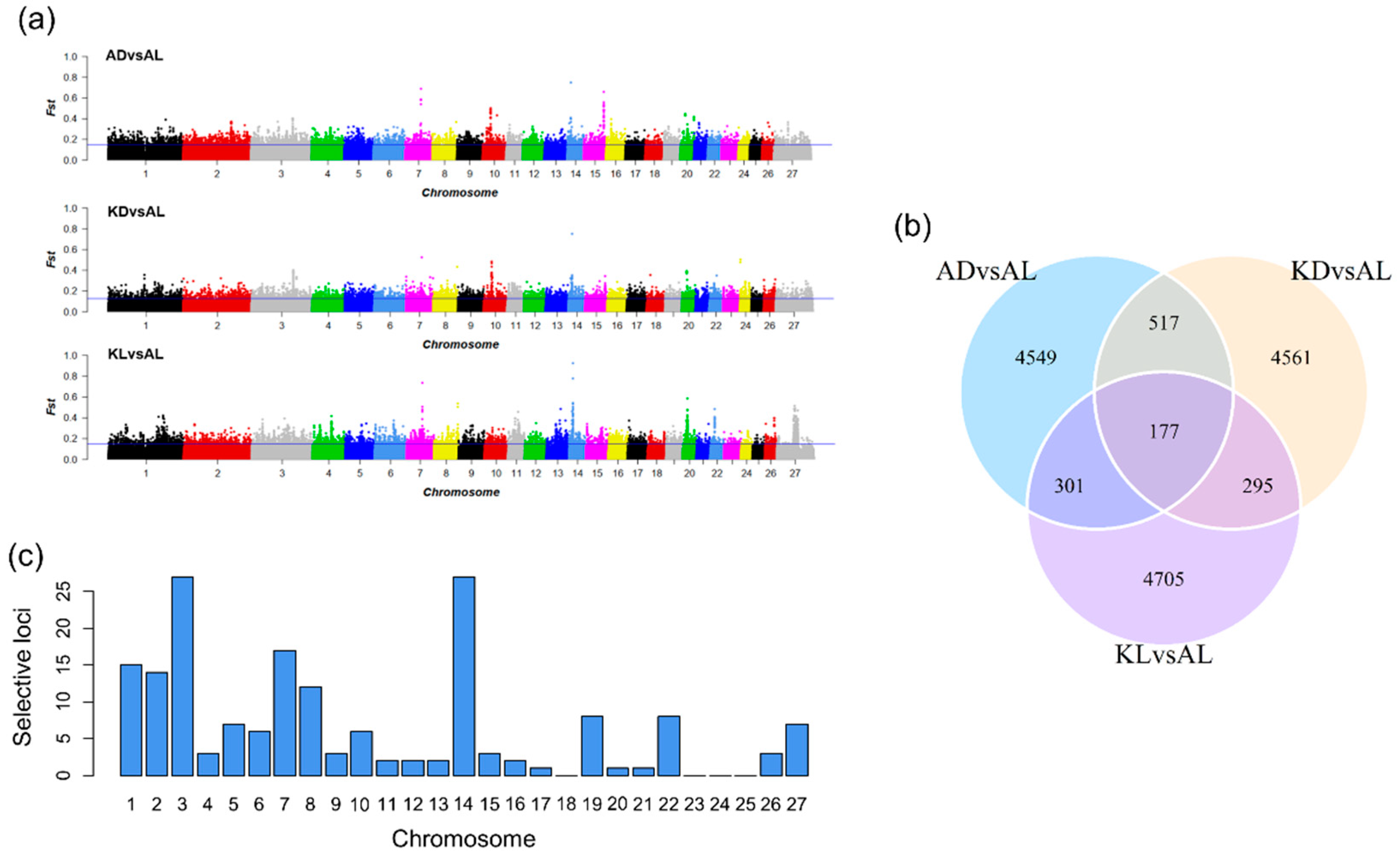
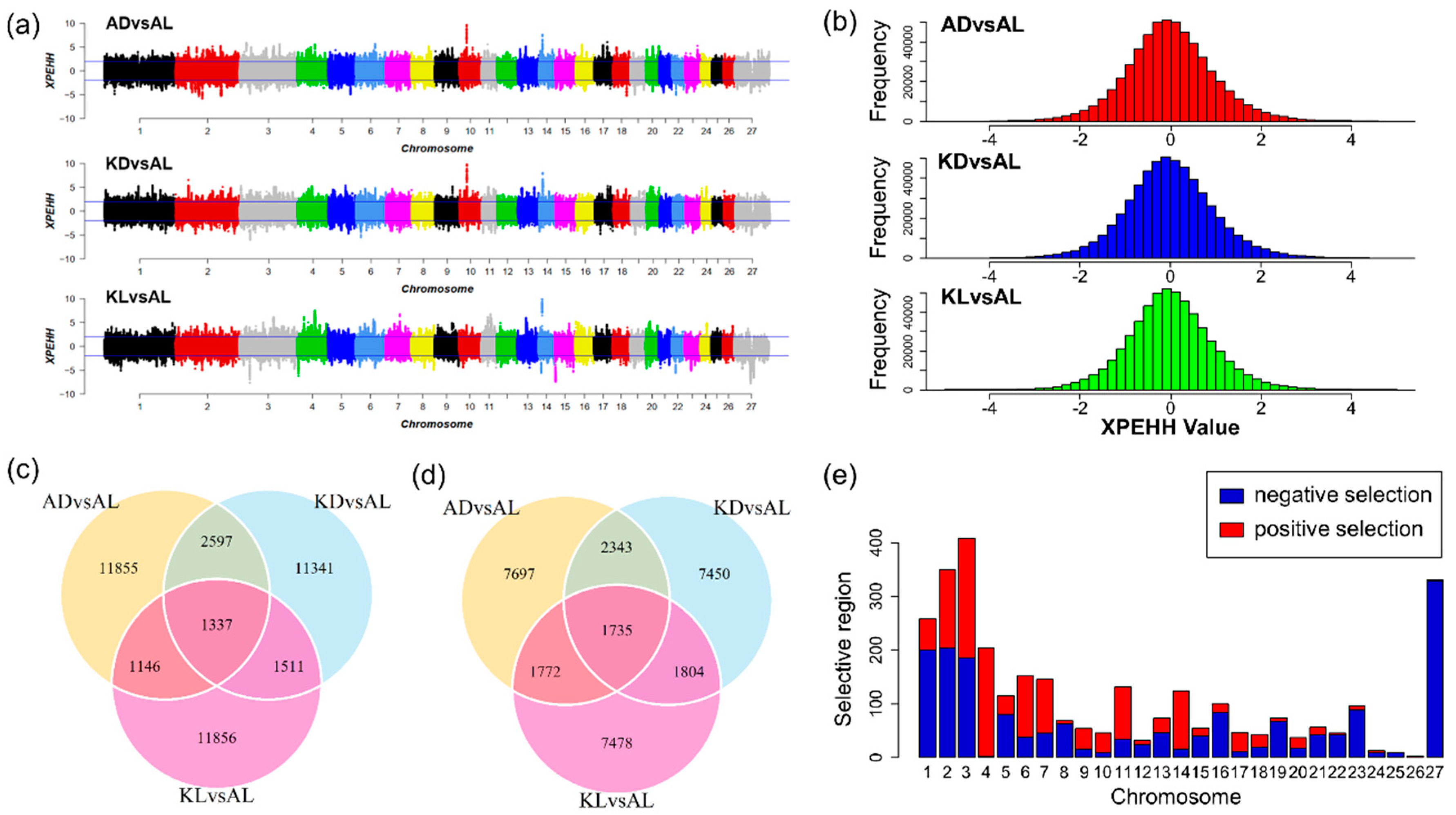
3.2. The Selection Signatures
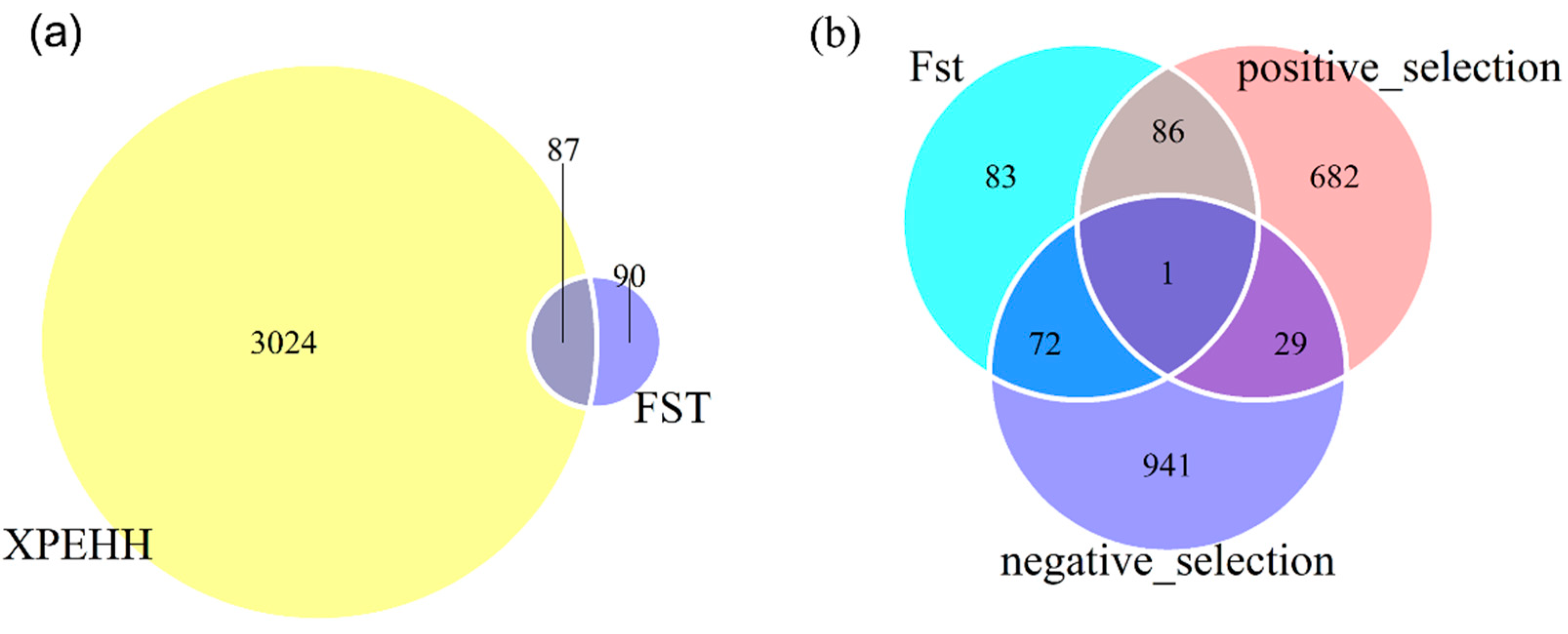
3.3. Functional Annotation
3.4. GO Term Enrichment Analysis
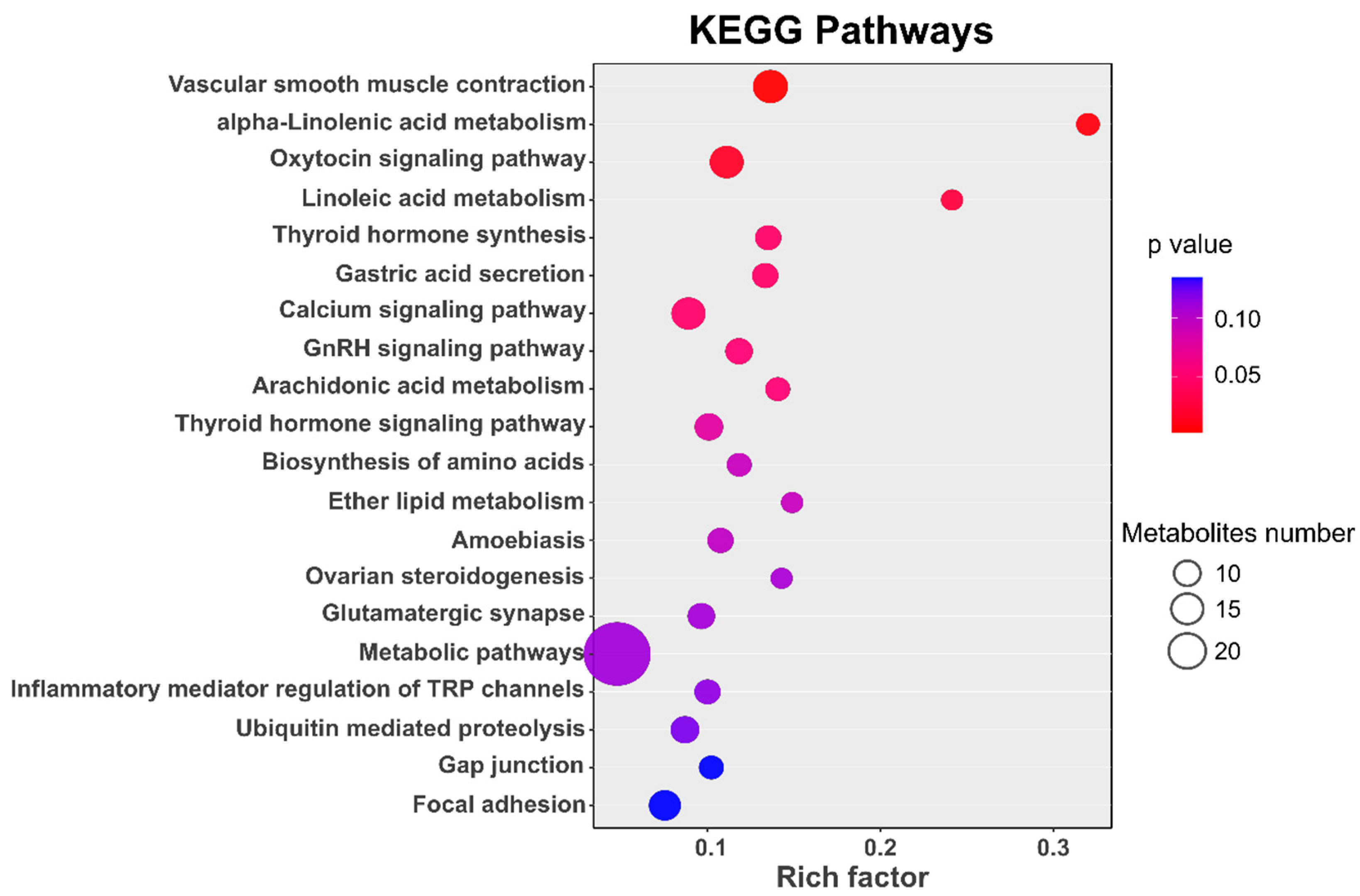
3.5. Pathway Enrichment Analysis
3.6. Candidate Genes Related to Litter Size
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A

References
- Tian, D.; Araki, H.; Stahl, E.; Bergelson, J.; Kreitman, M. Signature of balancing selection in Arabidopsis. Proc. Natl. Acad. Sci. USA 2002, 99, 11525–11530. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Georges, M. Domestic-animal genomics: Deciphering the genetics of complex traits. Nat. Rev. Genet. 2004, 5, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-J.; Yang, J.; Xie, X.-L.; Lv, F.-H.; Cao, Y.-H.; Li, W.-R.; Liu, M.-J.; Wang, Y.-T.; Li, J.-Q.; Liu, Y.-G.; et al. The Genome Landscape of Tibetan Sheep Reveals Adaptive Introgression from Argali and the History of Early Human Settlements on the Qinghai–Tibetan Plateau. Mol. Boil. Evol. 2018, 36, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, W.; Li, F.; Yue, X. Selection signature analysis reveals genes underlying sheep milking performance. Arch. Anim. Breed. 2019, 62, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Sabeti, P.; He, Y.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Mulsant, P.; Lecerf, F.; Fabre, S.; Schibler, L.; Monget, P.; Lanneluc, I.; Pisselet, C.; Riquet, J.; Monniaux, D.; Callebaut, I.; et al. Mutation in bone morphogenetic protein receptor-IB is associated with increased ovulation rate in Booroola Merino ewes. Proc. Natl. Acad. Sci. USA 2001, 98, 5104–5109. [Google Scholar] [CrossRef]
- Yue, G.H. Reproductive characteristics of Chinese Hu sheep. Anim. Reprod. Sci. 1996, 44, 223–230. [Google Scholar] [CrossRef]
- Li, M.-H.; Xu, S.-S. Recent advances in understanding genetic variants associated with economically important traits in sheep (Ovis aries) revealed by high-throughput screening technologies. Front. Agric. Sci. Eng. 2017, 4, 279. [Google Scholar]
- Galloway, S.M.; McNatty, K.P.; Cambridge, L.M.; Laitinen, M.P.; Juengel, J.; Jokiranta, T.S.; McLaren, R.J.; Luiro, K.; Dodds, K.; Montgomery, G.W.; et al. Mutations in an oocyte-derived growth factor gene (BMP15) cause increased ovulation rate and infertility in a dosage-sensitive manner. Nat. Genet. 2000, 25, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Våge, D.I.; Husdal, M.; Kent, M.P.; Klemetsdal, G.; Boman, I.A. A missense mutation in growth differentiation factor 9 (GDF9) is strongly associated with litter size in sheep. BMC Genet. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Ovarian transcriptomic study reveals the differential regulation of miRNAs and lncRNAs related to fecundity in different sheep. Sci. Rep. 2016, 6, 35299. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Pemberton, J.M.; Pilkington, J.G.; Clutton-Brock, T.H.; Kruuk, L.E. Trading offspring size for number in a variable environment: Selection on reproductive investment in female Soay sheep. J. Anim. Ecol. 2009, 78, 354–364. [Google Scholar] [CrossRef]
- Li, X.L.; Gong, Y.F.; Zhang, J.W.; Liu, Z.Z.; Valentini, A. Study on polymorphisms of microsatellites DNA of six Chinese indigenous sheep breeds. Acta Genet. Sin. 2004, 31, 1203–1210. [Google Scholar]
- Zheng, P.; Zhang, Z.; Chen, X.; Tu, Y. Sheep and Goat Breeds in China; Shanghai Scientific & Technical Publishers: Shanghai, China, 1988; pp. 38–40. [Google Scholar]
- Porter, V. Mason’s World Dictionary of Livestock Breeds, Types and Varieties, 5th ed.; CAB International: Wallingford, UK, 2002; pp. 58–64. [Google Scholar]
- Purcell, S.M.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.R.; Albers, C.A.; Banks, E.; Depristo, M.A.; Ha, G.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Delaneau, O.; Marchini, J. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun. 2014, 5, 3934. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Hernandez, R.D. selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L.; Clark, A.G.; Clark, A.G. Principles of Population Genetics; Sinauer associates: Sunderland, MA, USA, 1997. [Google Scholar]
- Thrun, L.A.; Dahl, G.E.; Evans, N.P.; Karsch, F.J. A critical period for thyroid hormone action on seasonal changes in reproductive neuroendocrine function in the ewe. Endocrinology 1997, 138, 3402–3409. [Google Scholar] [CrossRef] [PubMed]
- Bagheripuor, F.; Ghanbari, M.; Piryaei, A.; Ghasemi, A. Effects of fetal hypothyroidism on uterine smooth muscle contraction and structure of offspring rats. Exp. Physiol. 2018, 103, 683–692. [Google Scholar] [CrossRef]
- Vastagh, C.; Rodolosse, A.; Solymosi, N.; Liposits, Z. Altered Expression of Genes Encoding Neurotransmitter Receptors in GnRH Neurons of Proestrous Mice. Front. Cell. Neurosci. 2016, 10, 230. [Google Scholar] [CrossRef]
- Rekik, M.; Haile, A.; Abebe, A.; Muluneh, D.; Goshme, S.; Ben Salem, I.; Hilali, M.E.-D.; Lassoued, N.; Chanyalew, Y.; Rischkowsky, B. GnRH and prostaglandin-based synchronization protocols as alternatives to progestogen-based treatments in sheep. Reprod. Domest. Anim. 2016, 51, 924–929. [Google Scholar] [CrossRef]
- Jung, D.; Xiong, J.; Ye, M.; Qin, X.; Li, L.; Cheng, S.; Luo, M.; Peng, J.; Dong, J.; Tang, F.; et al. In vitro differentiation of human embryonic stem cells into ovarian follicle-like cells. Nat. Commun. 2017, 8, 15680. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Chen, Q.; Zhang, X.; Wang, Q.; Pan, Y. A genome scan for selection signatures in Taihu pig breeds using next-generation sequencing. Animal 2019, 13, 683–693. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Liu, X.T.; DeSpain, K. Pregnancy modifies the large conductance Ca2+-activated K+ channel and cGMP-dependent signaling pathway in uterine vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1878–H1887. [Google Scholar] [CrossRef]
- Iwama, N.; Metoki, H.; Nishigori, H.; Mizuno, S.; Takahashi, F.; Tanaka, K.; Watanabe, Z.; Saito, M.; Sakurai, K.; Ishikuro, M.; et al. Blood pressure changes during twin pregnancies: The Japan Environment and Children’s Study. J. Hypertens. 2019, 37, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Tanbe, A.F.; Khalil, R.A. Circulating and Vascular Bioactive Factors during Hypertension in Pregnancy. Curr. Bioact. Compd. 2010, 6, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Foroughinia, G.; Fazileh, A.; Eghbalsaied, S. Expression of genes involved in BMP and estrogen signaling and AMPK production can be important factors affecting total number of antral follicles in ewes. Theriogenology 2017, 91, 36–43. [Google Scholar] [CrossRef]
- Ozmen, O.; Seker, I.; Kul, B.C.; Ertugrul, O. Haplotype variation of estrogen receptor-alpha (ER-alpha) gene exon 4 in Turkish sheep breeds. Genetika 2012, 48, 1185–1189. [Google Scholar] [PubMed]
- Gratao, A.A.; Dahlhoff, M.; Sinowatz, F.; Wolf, E.; Schneider, M.R. Betacellulin overexpression in the mouse ovary leads to MAPK3/MAPK1 hyperactivation and reduces litter size by impairing fertilization. Biol. Reprod. 2008, 78, 43–52. [Google Scholar] [CrossRef]
- Macháty, Z.; Funahashi, H.; Day, B.N.; Prather, R.S. Developmental changes in the intracellular Ca2+ release mechanisms in porcine oocytes. Biol. Reprod. 1997, 56, 921–930. [Google Scholar] [CrossRef]
- Cechová, M.; Wolf, J.; Trcka, P. Impact of RYR1 genotype of Piétrain boars on litter traits of Czech Large White x Czech Landrace crossbred sows. J. Anim. Breed. Genet. 2007, 124, 86–93. [Google Scholar] [CrossRef]
- Yuan, Y.; Wheeler, M.B.; Krisher, R.L. Disrupted redox homeostasis and aberrant redox gene expression in porcine oocytes contribute to decreased developmental competence. Biol. Reprod. 2012, 87, 78. [Google Scholar] [CrossRef]
- Liu, C.; Ran, X.-Q.; Niu, X.; Li, S.; Wang, J.; Zhang, Q. Insertion of 275-bp SINE into first intron of PDIA4 gene is associated with litter size in Xiang pigs. Anim. Reprod. Sci. 2018, 195, 16–23. [Google Scholar] [CrossRef]
- Vargas-Pinilla, P.; Paixão-Côrtes, V.R.; Paré, P.; Tovo-Rodrigues, L.; Vieira, C.M.D.A.G.; Xavier, A.; Comas, D.; Pissinatti, A.; Sinigaglia, M.; Rigo, M.M.; et al. Evolutionary pattern in the OXT-OXTR system in primates: Coevolution and positive selection footprints. Proc. Natl. Acad. Sci. USA 2015, 112, 88–93. [Google Scholar] [CrossRef]
- Gimpl, G.; Fahrenholz, F. The oxytocin receptor system: Structure, function, and regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef] [PubMed]
- Gysin, J.; Pouvelle, B.; le Tonquèze, M.; Edelman, L.; Boffa, M.C. Chondroitin sulfate of thrombomodulin is an adhesion receptor for Plasmodium falciparum-infected erythrocytes. Mol. Biochem. Parasitol. 1997, 88, 267–271. [Google Scholar] [CrossRef]
- Luo, W.; Wiltbank, M.C. Distinct regulation by steroids of messenger RNAs for FSHR and CYP19A1 in bovine granulosa cells. Biol. Reprod. 2006, 75, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Garverick, H.A. Expression of steroidogenic enzyme and gonadotropin receptor genes in bovine follicles during ovarian follicular waves: A review. J. Anim. Sci. 1998, 76, 1903–1921. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, Q.; Wang, X. Expression of CYP11A1, CYP17A1 and CYP19A1 genes in hypothalamus-pituitary-ovary axis of Small Tail Han sheep. J. China Agric. Univ. 2019, 24, 87–93. [Google Scholar]
- Fabre, S.; Pierre, A.; Mulsant, P.; Bodin, L.; Di Pasquale, E.; Persani, L.; Monget, P.; Monniaux, D. Regulation of ovulation rate in mammals: Contribution of sheep genetic models. Reprod. Biol. Endocrinol. 2006, 4, 20. [Google Scholar] [CrossRef]
- McNatty, K.P.; Moore, L.G.; Hudson, N.L.; Quirke, L.D.; Lawrence, S.B.; Reader, K.; Hanrahan, J.P.; Smith, P.; Groome, N.P.; Laitinen, M.; et al. The oocyte and its role in regulating ovulation rate: A new paradigm in reproductive biology. Reproduction 2004, 128, 379–386. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed Pair 1 | The NO. of SNP | Fst | XPEHH+ 2 | XPEHH− 3 | |||
|---|---|---|---|---|---|---|---|
| Selective SNPs (Threshold Value) | Regions | Core SNPs | Regions | Core SNPs | Selective Regions | ||
| AD-AL | 542516 | 5545 (0.15) | 1705 | 16,935 | 1399 | 13,547 | 1416 |
| KD-AL | 543134 | 5550 (0.13) | 1716 | 16,786 | 1409 | 13,332 | 1471 |
| KL-AL | 536778 | 5479 (0.15) | 1404 | 15,850 | 1203 | 12,789 | 1314 |
| Term | Database | ID | No. of Genes | p-Value | Corrected p-Value 1 | Genes |
|---|---|---|---|---|---|---|
| Multicellular organism reproduction | Gene Ontology | GO: 0032504 | 12 | 0.002209 | 0.109913 | ESR1|OXTR|STAT5B|CATSPERG|GGN|SIRT2| TDRD7|MEA1|SLC26A3|SMAD5|PDGFRA|BBS4 |
| Oxytocin signaling pathway | KEGG PATHWAY | hsa04921 | 17 | 3.79 × 10−5 | 0.01319 | ADCY5|OXTR|MYLK|GNAS|MYLK3|RYR1|MAPK1| MYLK4|EEF2K|CAMK2B|PRKAG2|CALML4| PLA2G4E|PLA2G4D|PLA2G4F|NFATC2|PPP3R1 |
| Thyroid hormone synthesis | KEGG PATHWAY | hsa04918 | 10 | 0.000367 | 0.045264 | ADCY5|ASGR2|ASGR1|ATP1B2|GNAS|GPX8|PAX8| SLC26A4|PDIA4|ATP1A3 |
| GnRH signaling pathway | KEGG PATHWAY | hsa04912 | 11 | 0.000534 | 0.050691 | ADCY5|GNAS|MAPK1|PRKCD|CAMK2B|GNA11| EGR1|CALML4|PLA2G4E|PLA2G4D|PLA2G4F |
| Thyroid hormone signaling pathway | KEGG PATHWAY | hsa04919 | 12 | 0.001109 | 0.077681 | MED12L|ATP1B2|TP53|KAT2A|MAPK1|ITGAV| PLCE1|TSC2|DIO2|ESR1|GSK3B|ATP1A3 |
| Ovarian steroidogenesis | KEGG PATHWAY | hsa04913 | 7 | 0.002069 | 0.107993 | ADCY5|GNAS|PLA2G4E|PLA2G4D|PLA2G4F| CYP19A1|HSD17B2 |
| Candidate Genes | GO Terms and KEGG Pathways | Test 2 | |||
|---|---|---|---|---|---|
| Term | ID | p-Value | Corrected p-Value 1 | ||
| ESR1 | multicellular organism reproduction | GO: 0032504 | 0.002209 | 0.109913 | XPEHH |
| Thyroid hormone signaling pathway | hsa04919 | 0.001109 | 0.077681 | ||
| OXTR | multicellular organism reproduction | GO: 0032504 | 0.002209 | 0.109913 | Fst/XPEHH |
| Oxytocin signaling pathway | hsa04921 | 3.79 × 10−5 | 0.01319 | ||
| MAPK1 | Oxytocin signaling pathway | hsa04921 | 3.79 × 10−5 | 0.01319 | XPEHH |
| GnRH signaling pathway | hsa04912 | 0.000534 | 0.050691 | ||
| Thyroid hormone signaling pathway | hsa04919 | 0.001109 | 0.077681 | ||
| RYR1 | Oxytocin signaling pathway | hsa04921 | 3.79 × 10−5 | 0.01319 | XPEHH |
| PDIA1 | Thyroid hormone synthesis | hsa04919 | 0.000367 | 0.045264 | XPEHH |
| CYP19A1 | Ovarian steroidogenesis | hsa04913 | 0.0020688 | 0.1079928 | Fst/XPEHH |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Niu, Z.; Zeng, Z.; Jiang, Y.; Jiang, Y.; Ding, Y.; Tang, S.; Shi, H.; Ding, X. Using High-Density SNP Array to Reveal Selection Signatures Related to Prolificacy in Chinese and Kazakhstan Sheep Breeds. Animals 2020, 10, 1633. https://doi.org/10.3390/ani10091633
Wang Y, Niu Z, Zeng Z, Jiang Y, Jiang Y, Ding Y, Tang S, Shi H, Ding X. Using High-Density SNP Array to Reveal Selection Signatures Related to Prolificacy in Chinese and Kazakhstan Sheep Breeds. Animals. 2020; 10(9):1633. https://doi.org/10.3390/ani10091633
Chicago/Turabian StyleWang, Yi, Zhigang Niu, Zhengcheng Zeng, Yao Jiang, Yifan Jiang, Yugong Ding, Sen Tang, Hongcai Shi, and Xiangdong Ding. 2020. "Using High-Density SNP Array to Reveal Selection Signatures Related to Prolificacy in Chinese and Kazakhstan Sheep Breeds" Animals 10, no. 9: 1633. https://doi.org/10.3390/ani10091633
APA StyleWang, Y., Niu, Z., Zeng, Z., Jiang, Y., Jiang, Y., Ding, Y., Tang, S., Shi, H., & Ding, X. (2020). Using High-Density SNP Array to Reveal Selection Signatures Related to Prolificacy in Chinese and Kazakhstan Sheep Breeds. Animals, 10(9), 1633. https://doi.org/10.3390/ani10091633