
Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents and Microbiological Media
2.2. Microbial Media and Cultures
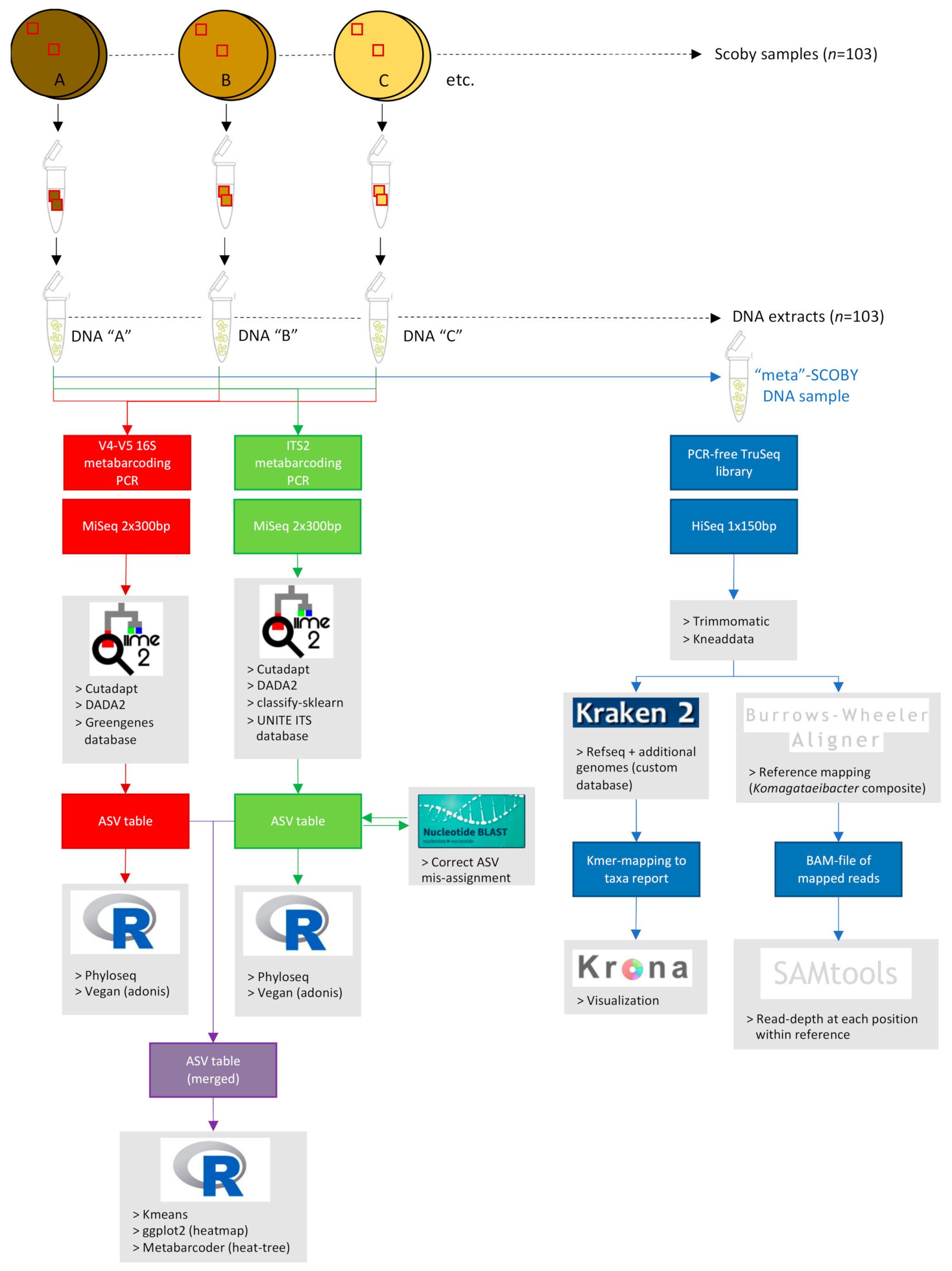
2.3. Kombucha SCOBY Starter Culture Sampling and Processing
2.3.1. Sectioning of Commercial Kombucha SCOBY (Spatial Analysis Study)
2.3.2. Collection of Representative Commercial Kombucha SCOBY (Taxonomic Diversity Study)
2.3.3. Sample Homogenization and DNA Extraction
2.4. Quantitive PCR Estimation of Kombucha SCOBY Microbial Population Size
2.5. Metabarcoding Analyses of Kombucha SCOBY Bacterial and Fungal Communities
2.5.1. Metabarcoding Library Preparation and Sequencing
2.5.2. Metabarcoding Sequence Processing and Analyses
2.6. Shotgun Metagenomic Sequencing Analysis of Composite ‘Meta-’SCOBY DNA Sample
2.6.1. Meta-SCOBY Library Preparation and Sequencing
2.6.2. Sequence Pre-Processing
2.6.3. Kmer Analysis of Meta-SCOBY Community Composition
2.6.4. Mapping of Meta-SCOBY Reads against a Komagataeibacter Composite Reference Genome
2.7. Statistical Analyses
2.8. Sequence Data Availability
3. Results
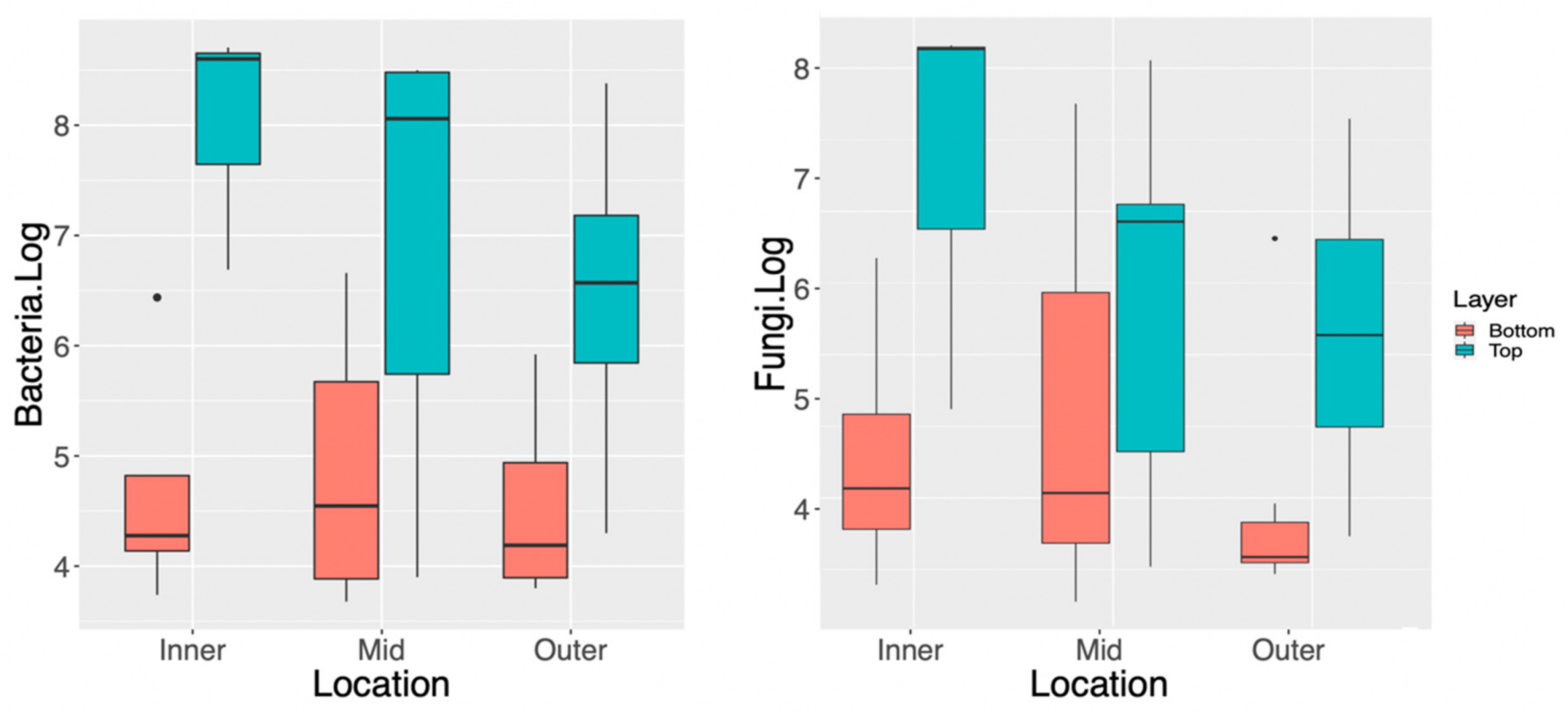
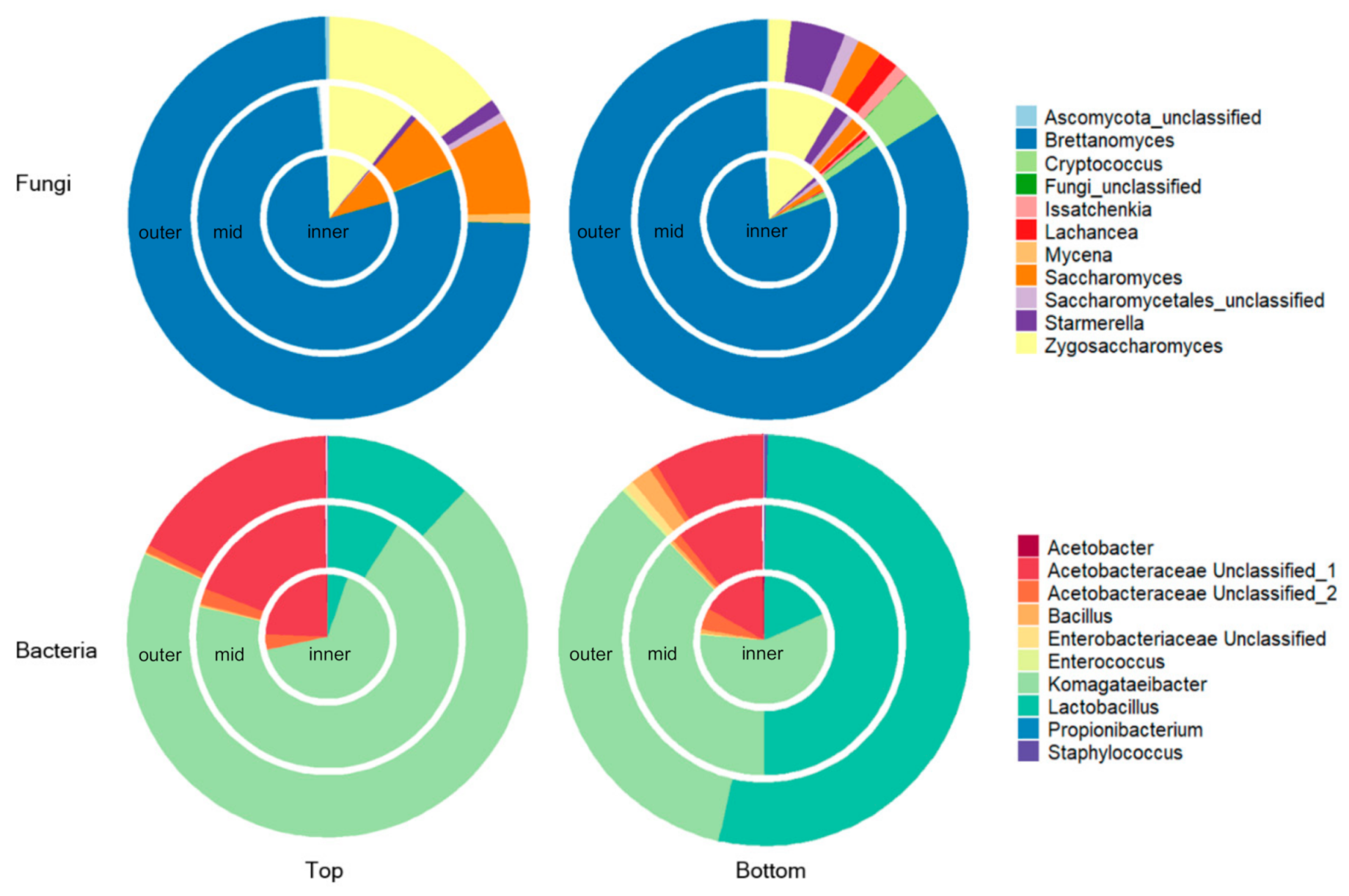
3.1. Spatial Distribution of Fungi and Bacteria within a Single Kombucha SCOBY
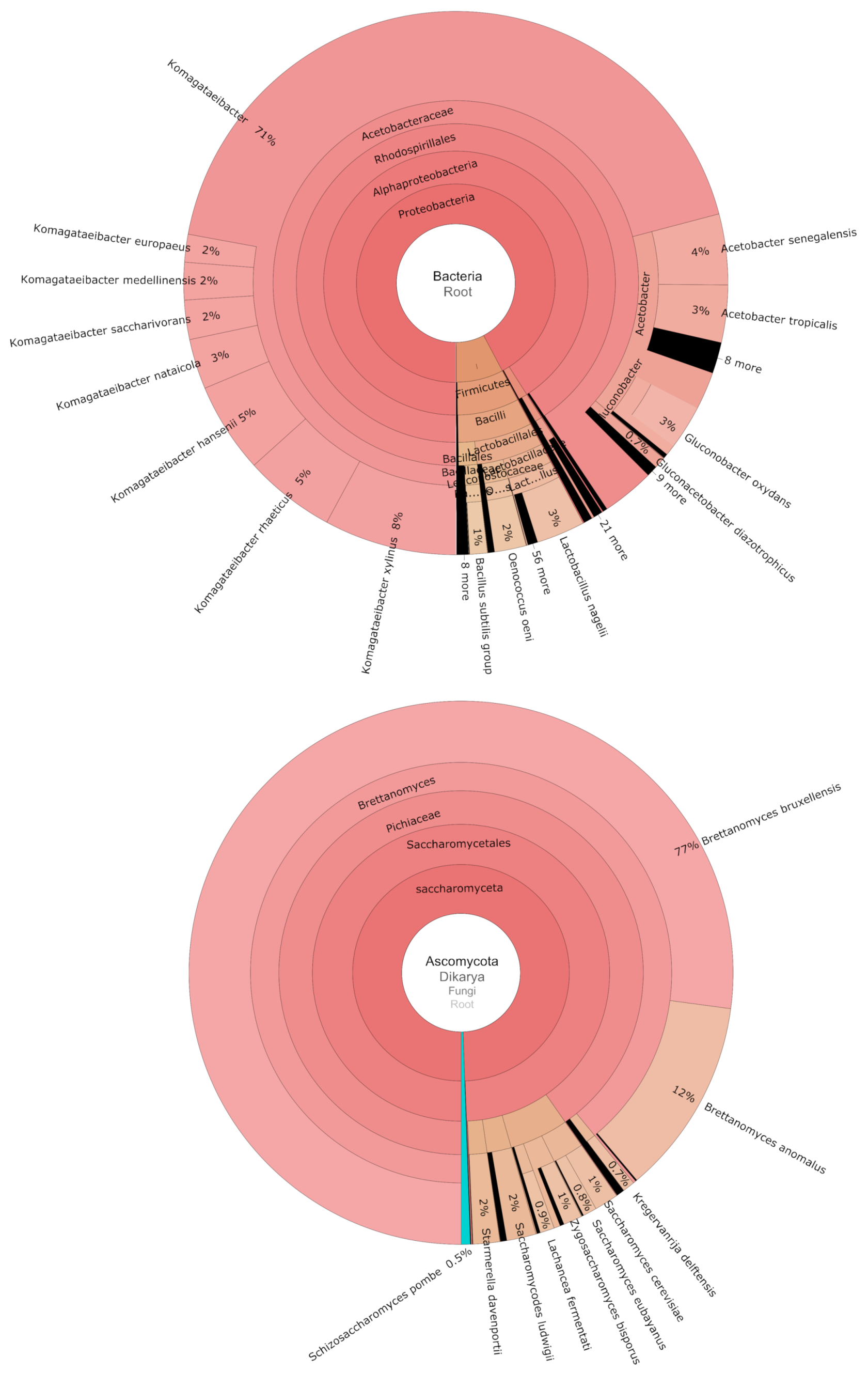
3.2. Fungal and Bacterial Communities Associated with 103 Commercial Kombucha SCOBY
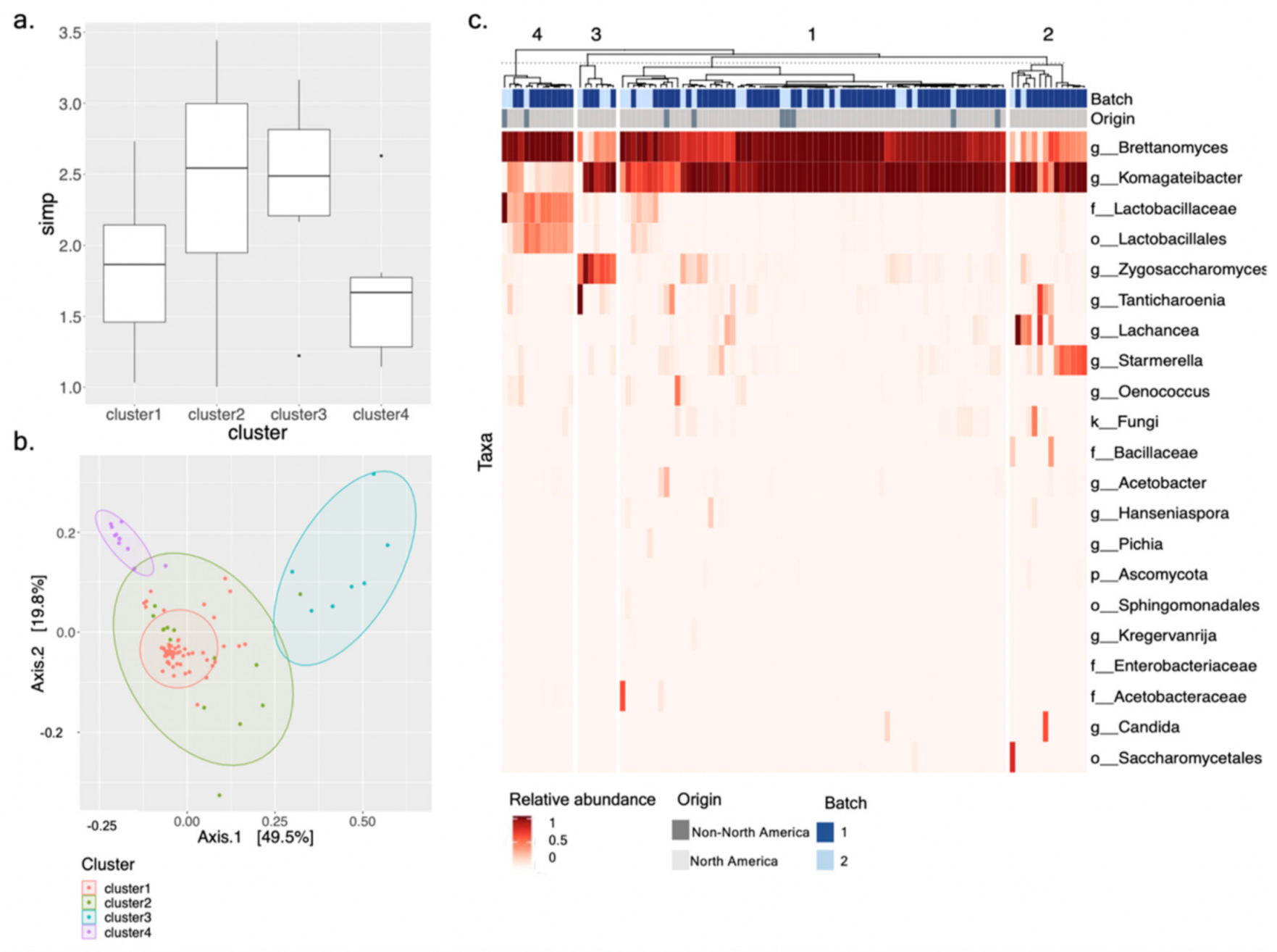
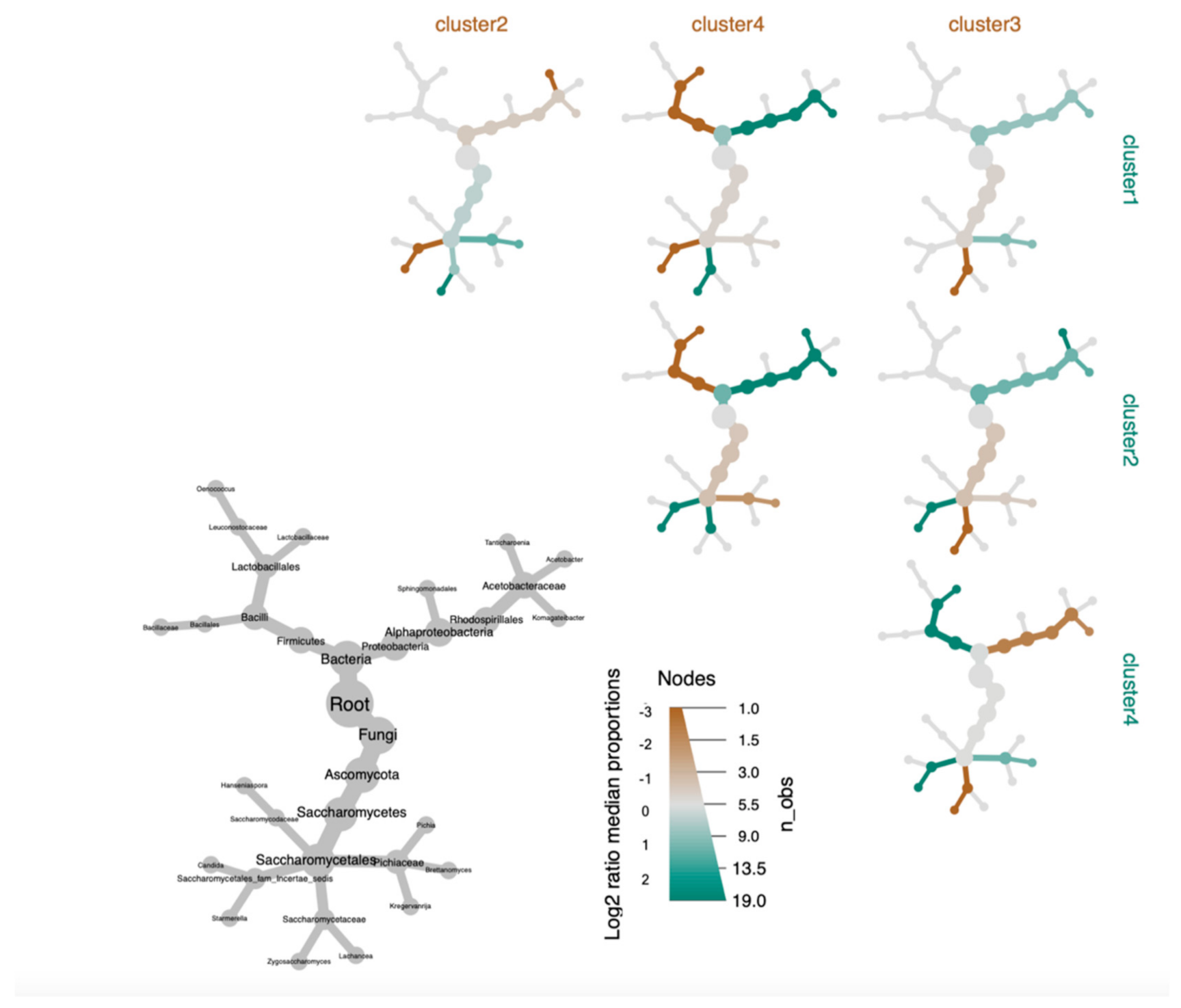
3.3. Defining Kombucha SCOBY Archetypes Based upon Microbial Community Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teoh, A.L.; Heard, G.; Cox, J. Yeast ecology of Kombucha fermentation. Int. J. Food Microbiol. 2004, 95, 119–126. [Google Scholar] [CrossRef]
- Malbaša, R.V.; Lončar, E.S.; Vitas, J.S.; Čanadanović-Brunet, J.M. Influence of starter cultures on the antioxidant activity of kombucha beverage. Food Chem. 2011, 127, 1727–1731. [Google Scholar] [CrossRef]
- Yamada, Y.; Yukphan, P.; Vu, H.T.L.; Muramatsu, Y.; Ochaikul, D.; Nakagawa, Y. Subdivision of the Genus Gluconacetobacter Yamada, Hoshino and Ishikawa 1998: The Proposal of Komagataeibacter Gen. Nov., for Strains Accommodated to the Gluconacetobacter Xylinus Group in the α-Proteobacteria. Ann. Microbiol. 2012, 62, 849–859. [Google Scholar] [CrossRef]
- Reva, O.N.; Zaets, I.E.; Ovcharenko, L.P.; Kukharenko, O.E.; Shpylova, S.P.; Podolich, O.V.; de Vera, J.-P.; Kozyrovska, N.O. Metabarcoding of the kombucha microbial community grown in different microenvironments. AMB Express 2015, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Troise, A.D.; Vitaglione, P.; Ercolini, D. Different temperatures select distinctive acetic acid bacteria species and promotes organic acids production during kombucha tea fermentation. Food Microbiol. 2018, 73, 11–16. [Google Scholar] [CrossRef]
- Machado, R.T.A.; Gutierrez, J.; Tercjak, A.; Trovatti, E.; Uahib, F.G.M.; de Moreno, G.P.; Nascimento, A.P.; Berreta, A.A.; Ribeiro, S.J.L.; Barud, H.S. komagataeibacter rhaeticus as an alternative bacteria for cellulose production. Carbohydr. Polym. 2016, 152, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Semjonovs, P.; Ruklisha, M.; Paegle, L.; Saka, M.; Treimane, R.; Skute, M.; Rozenberga, L.; Vikele, L.; Sabovics, M.; Cleenwerck, I. Cellulose synthesis by Komagataeibacter rhaeticus strain P 1463 isolated from Kombucha. Appl. Microbiol. Biotechnol. 2017, 101, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Gaggìa, F.; Baffoni, L.; Galiano, M.; Nielsen, D.S.; Jakobsen, R.R.; Castro-Mejía, J.L.; Bosi, S.; Truzzi, F.; Musumeci, F.; Dinelli, G.; et al. Kombucha Beverage from Green, Black and Rooibos Teas: A Comparative Study Looking at Microbiology, Chemistry and Antioxidant Activity. Nutrients 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, R.A.C.; Berretta, A.A.; Barud, H.d.S.; Ribeiro, S.J.L.; González-García, L.N.; Zucchi, T.D.; Goldman, G.H.; Riaño-Pachón, D.M. Draft Genome Sequence of Komagataeibacter Intermedius Strain AF2, a Producer of Cellulose, Isolated from Kombucha Tea. Genome Announc. 2015, 3, 6. [Google Scholar] [CrossRef]
- Sievers, M.; Lanini, C.; Weber, A.; Schuler-Schmid, U.; Teuber, M. Microbiology and Fermentation Balance in a Kombucha Beverage Obtained from a Tea Fungus Fermentation. Syst. Appl. Microbiol. 1995, 18, 590–594. [Google Scholar] [CrossRef]
- Blanc, P.J. Characterization of the tea fungus metabolites. Biotechnol. Lett. 1996, 18, 139–142. [Google Scholar] [CrossRef]
- Balentine, D.A.; Wiseman, S.A.; Bouwens, L.C. The chemistry of tea flavonoids. Crit. Rev. Food Sci. Nutr. 1997, 37, 693–704. [Google Scholar] [CrossRef]
- Liu, C.-H.; Hsu, W.-H.; Lee, F.-L.; Liao, C.-C. The isolation and identification of microbes from a fermented tea beverage, Haipao, and their interactions during Haipao fermentation. Food Microbiol. 1996, 13, 407–415. [Google Scholar] [CrossRef]
- Marsh, A.J.; O’Sullivan, O.; Hill, C.; Ross, R.P.; Cotter, P.D. Sequence-based analysis of the bacterial and fungal compositions of multiple kombucha (tea fungus) samples. Food Microbiol. 2014, 38, 171–178. [Google Scholar] [CrossRef]
- Chen, C.; Liu, B.Y. Changes in major components of tea fungus metabolites during prolonged fermentation. J. Appl. Microbiol. 2000, 89, 834–839. [Google Scholar] [CrossRef]
- Coton, M.; Pawtowski, A.; Taminiau, B.; Burgaud, G.; Deniel, F.; Coulloumme-Labarthe, L.; Fall, A.; Daube, G.; Coton, E. Unraveling microbial ecology of industrial-scale Kombucha fermentations by metabarcoding and culture-based methods. FEMS Microbiol. Ecol. 2017, 93, 048. [Google Scholar] [CrossRef]
- Chakravorty, S.; Bhattacharya, S.; Chatzinotas, A.; Chakraborty, W.; Bhattacharya, D.; Gachhui, R. Kombucha tea fermentation: Microbial and biochemical dynamics. Int. J. Food Microbiol. 2016, 220, 63–72. [Google Scholar] [CrossRef]
- Mayser, P.; Fromme, S.; Leitzmann, G.; Gründer, K. The yeast spectrum of the ‘tea fungus Kombucha’. Mycoses 1995, 38, 289–295. [Google Scholar] [CrossRef]
- Greenwalt, C.J.; Steinkraus, K.H.; Ledford, R.A. Kombucha, the Fermented Tea: Microbiology, Composition, and Claimed Health Effects. J. Food Prot. 2000, 63, 976–981. [Google Scholar] [CrossRef]
- Ben Taheur, F.; Mansour, C.; Ben Jeddou, K.; Machreki, Y.; Kouidhi, B.; Abdulhakim, J.A.; Chaieb, K. Aflatoxin B1 degradation by microorganisms isolated from Kombucha culture. Toxicon 2020, 179, 76–83. [Google Scholar] [CrossRef]
- Villarreal-Soto, S.A.; Beaufort, S.; Bouajila, J.; Souchard, J.-P.; Taillandier, P. Understanding Kombucha Tea Fermentation: A Review. J. Food Sci. 2018, 83, 580–588. [Google Scholar] [CrossRef]
- Tran, T.; Grandvalet, C.; Verdier, F.; Martin, A.; Alexandre, H.; Tourdot-Maréchal, R. Microbial Dynamics between Yeasts and Acetic Acid Bacteria in Kombucha: Impacts on the Chemical Composition of the Beverage. Foods 2020, 9, 963. [Google Scholar] [CrossRef]
- Pinto, L.; Malfeito-Ferreira, M.; Quintieri, L.; Silva, A.C.; Baruzzi, F. Growth and metabolite production of a grape sour rot yeast-bacterium consortium on different carbon sources. Int. J. Food Microbiol. 2019, 296, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese Rind Communities Provide Tractable Systems for in Situ and in Vitro Studies of Microbial Diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Paramithiotis, S.; Shin, H.-S. Kimchi and Other Widely Consumed Traditional Fermented Foods of Korea: A Review. Front. Microbiol. 2016, 7, 1493. [Google Scholar] [CrossRef] [PubMed]
- Filippis, F.D.; Parente, E.; Ercolini, D. Metagenomics insights into food fermentations. Microb. Biotechnol. 2017, 10, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A.C.; Nello, A.V.; Ernst, R.K.; Schertzer, J.W. Analysis of Pseudomonas Aeruginosa Biofilm Membrane Vesicles Supports Multiple Mechanisms of Biogenesis. PLoS ONE 2019, 14, e0212275. [Google Scholar] [CrossRef]
- Unban, K.; Khatthongngam, N.; Pattananandecha, T.; Saenjum, C.; Shetty, K.; Khanongnuch, C. Microbial Community Dynamics During the Non-Filamentous Fungi Growth-Based Fermentation Process of Miang, a Traditional Fermented Tea of North Thailand and Their Product Characterizations. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- De Gregoris, T.B.; Aldred, N.; Clare, A.S.; Burgess, J.G. Improvement of Phylum- and Class-Specific Primers for Real-Time PCR Quantification of Bacterial Taxa. J. Microbiol. Methods 2011, 86, 351–356. [Google Scholar] [CrossRef]
- Hierro, N.; Esteve-Zarzoso, B.; González, A.; Mas, A.; Guillamón, J.M. Real-Time Quantitative PCR (QPCR) and Reverse Transcription-QPCR for Detection and Enumeration of Total Yeasts in Wine. Appl. Environ. Microbiol. 2006, 72, 7148–7155. [Google Scholar] [CrossRef]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Mills, D.A. Improved Selection of Internal Transcribed Spacer-Specific Primers Enables Quantitative, Ultra-High-Throughput Profiling of Fungal Communities. Appl. Environ. Microbiol. 2013, 79, 2519–2526. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-Throughput Amplicon Sequencing of the Full-Length 16S RRNA Gene with Single-Nucleotide Resolution. Nucl. Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef]
- De Santis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi—Recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S rRNA Gene Analysis with QIIME2. In Microbiome Analysis: Methods and Protocols; Beiko, R.G., Hsiao, W., Parkinson, J., Eds.; Springer: New York, NY, USA, 2018; pp. 113–129. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Getting Started with ggplot2. In ggplot2: Elegant Graphics for Data Analysis; Wickham, H., Ed.; Springer: Cham, Switzerland, 2016; pp. 11–31. [Google Scholar]
- Lozupone, C.; Hamady, M.; Knight, R. UniFrac—An Online Tool for Comparing Microbial Community Diversity in a Phylogenetic Context. BMC Bioinf. 2006, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.; O’hara, R.B.; Simpson, G. Community Ecology Package. R Package Version 2.0. 2013. Available online: http://sortie-admin.readyhosting.com/lme/R%20Packages/vegan.pdf (accessed on 9 April 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Spitaels, F.; Wieme, A.D.; Janssens, M.; Aerts, M.; Van Landschoot, A.; De Vuyst, L.; Vandamme, P. The microbial diversity of an industrially produced lambic beer shares members of a traditionally produced one and reveals a core microbiota for lambic beer fermentation. Food Microbiol. 2015, 49, 23–32. [Google Scholar] [CrossRef]
- Spitaels, F.; Wieme, A.D.; Janssens, M.; Aerts, M.; Daniel, H.-M.; Landschoot, A.V.; Vuyst, L.D.; Vandamme, P. The Microbial Diversity of Traditional Spontaneously Fermented Lambic Beer. PLoS ONE 2014, 9, e95384. [Google Scholar] [CrossRef]
- Domizio, P.; Lencioni, L.; Ciani, M.; Di Blasi, S.; Pontremolesi, C.; Sabatelli, M.P. Spontaneous and inoculated yeast populations dynamics and their effect on organoleptic characters of vinsanto wine under different process conditions. Int. J. Food Microbiol. 2007, 115, 281–289. [Google Scholar] [CrossRef]
- Combina, M.; Elía, A.; Mercado, L.; Catania, C.; Ganga, A.; Martinez, C. Dynamics of indigenous yeast populations during spontaneous fermentation of wines from Mendoza, Argentina. Int. J. Food Microbiol. 2005, 99, 237–243. [Google Scholar] [CrossRef]
- Di Maro, E.; Ercolini, D.; Coppola, S. Yeast dynamics during spontaneous wine fermentation of the Catalanesca grape. Int. J. Food Microbiol. 2007, 117, 201–210. [Google Scholar] [CrossRef]
- Abriouel, H.; Martín-Platero, A.; Maqueda, M.; Valdivia, E.; Martínez-Bueno, M. Biodiversity of the microbial community in a Spanish farmhouse cheese as revealed by culture-dependent and culture-independent methods. Int. J. Food Microbiol. 2008, 127, 200–208. [Google Scholar] [CrossRef]
- Marino, M.; Maifreni, M.; Rondinini, G. Microbiological Characterization of Artisanal Montasio Cheese: Analysis of Its Indigenous Lactic Acid Bacteria. FEMS Microbiol. Lett. 2003, 229, 133–140. [Google Scholar] [CrossRef]
- Podolich, O.; Kukharenko, O.; Haidak, A.; Zaets, I.; Zaika, L.; Storozhuk, O.; Palchikovska, L.; Orlovska, I.; Reva, O.; Borisova, T.; et al. Multimicrobial Kombucha Culture Tolerates Mars-Like Conditions Simulated on Low Earth Orbit. Astrobiology 2019, 19, 183–196. [Google Scholar] [CrossRef]
- Villarreal-Soto, S.A.; Bouajila, J.; Pace, M.; Leech, J.; Cotter, P.D.; Souchard, J.-P.; Taillandier, P.; Beaufort, S. Metabolome-Microbiome Signatures in the Fermented Beverage, Kombucha. Int. J. Food Microbiol. 2020, 333, 108778. [Google Scholar] [CrossRef]
- Herald, P.J.; Zottola, E.A. Attachment of Listeria Monocytogenes to Stainless Steel Surfaces at Various Temperatures and pH Values. J. Food Sci. 1988, 53, 1549–1562. [Google Scholar] [CrossRef]
- De Roos, J.; Vandamme, P.; De Vuyst, L. Wort Substrate Consumption and Metabolite Production During Lambic Beer Fermentation and Maturation Explain the Successive Growth of Specific Bacterial and Yeast Species. Front. Microbiol. 2018, 9, 2763. [Google Scholar] [CrossRef]
- Gorski, L.; Palumbo, J.D.; Mandrell, R.E. Attachment of Listeria Monocytogenes to Radish Tissue is Dependent upon Temperature and Flagellar Motility. Appl. Environ. Microbiol. 2003, 69, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Moltz, A.G.; Martin, S.E. Formation of Biofilms by Listeria monocytogenes under Various Growth Conditions. J. Food Prot. 2005, 68, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Folsom, J.P.; Siragusa, G.R.; Frank, J.F. Formation of Biofilm at Different Nutrient Levels by Various Genotypes of Listeria monocytogenes. J. Food Prot. 2006, 69, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Uscanga, B.; François, J.M. A Study of the Yeast Cell Wall Composition and Structure in Response to Growth Conditions and Mode of Cultivation. Lett. Appl. Microbiol. 2003, 37, 268–274. [Google Scholar] [CrossRef]
- Molina-Ramírez, C.; Castro, M.; Osorio, M.; Torres-Taborda, M.; Gómez, B.; Zuluaga, R.; Gómez, C.; Gañán, P.; Rojas, O.J.; Castro, C. Effect of Different Carbon Sources on Bacterial Nanocellulose Production and Structure Using the Low pH Resistant Strain Komagataeibacter Medellinensis. Materials 2017, 10, 639. [Google Scholar] [CrossRef]
- Laureys, D.; Aerts, M.; Vandamme, P.; De Vuyst, L. Oxygen and diverse nutrients influence the water kefir fermentation process. Food Microbiol. 2018, 73, 351–361. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; Turroni, F.; Sinderen, D.; Ventura, M. A microbiome reality check: Limitations of in silico-based metagenomic approaches to study complex bacterial communities. Environ. Microbiol. Rep. 2019, 11, 840–847. [Google Scholar] [CrossRef]
- Sternes, P.R.; Lee, D.; Kutyna, D.R.; Borneman, A.R. A combined meta-barcoding and shotgun metagenomic analysis of spontaneous wine fermentation. GigaScience 2017, 6, 040. [Google Scholar] [CrossRef]
- Arıkan, M.; Mitchell, A.L.; Finn, R.D.; Gürel, F. Microbial composition of Kombucha determined using amplicon sequencing and shotgun metagenomics. J. Food Sci. 2020, 85, 455–464. [Google Scholar] [CrossRef]
- Csoma, H.; Sipiczki, M. Taxonomic Reclassification of Candida Stellata Strains Reveals Frequent Occurrence of Candida Zemplinina in Wine Fermentation. FEMS Yeast Res. 2008, 8, 328–336. [Google Scholar] [CrossRef]
- Tu, C.; Hu, W.; Tang, S.; Meng, L.; Huang, Z.; Xu, X.; Xia, X.; Azi, F.; Dong, M. Isolation and identification of Starmerella davenportii strain Do18 and its application in black tea beverage fermentation. Food Sci. Hum. Wellness 2020, 9, 355–362. [Google Scholar] [CrossRef]
- Chen, C.; Liu, B. Studies in Microbiological Quality and Survival of Candida Albicans in the Tea Fungi. J. Agric. Forest. 1997, 46, 53–64. [Google Scholar]
- Hesseltine, C.W. A Millennium of Fungi, Food, and Fermentation. Mycologia 1965, 57, 149–197. [Google Scholar] [CrossRef]
- Watawana, M.; Jayawardena, N.; Gunawardhana, C.; Viduranga, Y. Health, Wellness, and Safety Aspects of the Consumption of Kombucha. J. Chem. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef]
- Shayevitz, A.; Harrison, K.; Curtin, C. Barrel-Induced Variation in the Microbiome and Mycobiome of Aged Sour Ale and Imperial Porter Beer. J. Am. Soc. Brew. Chem. 2020, 79, 1–8. [Google Scholar] [CrossRef]
- Muhammad, M.H.; Idris, A.L.; Fan, X.; Guo, Y.; Yu, Y.; Jin, X.; Qiu, J.; Guan, X.; Huang, T. Beyond Risk: Bacterial Biofilms and Their Regulating Approaches. Front. Microbiol. 2020, 11, 928. [Google Scholar] [CrossRef]
- Dang, H.; Lovell, C.R. Microbial Surface Colonization and Biofilm Development in Marine Environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef]
- Landis, E.A.; Oliverio, A.M.; McKenney, E.A.; Nichols, L.M.; Kfoury, N.; Biango-Daniels, M.; Shell, L.K.; Madden, A.A.; Shapiro, L.; Sakunala, S.; et al. The diversity and function of sourdough starter microbiomes. eLife 2021, 10, e61644. [Google Scholar] [CrossRef]
- Drysdale, G.S.; Fleet, G.H. The Growth and Survival of Acetic Acid Bacteria in Wines at Different Concentrations of Oxygen. Am. J. Enol. Vitic. 1989, 40, 99–105. [Google Scholar]
- Du Toit, W.; Pretorius, I.; Lonvaud-Funel, A. The effect of sulphur dioxide and oxygen on the viability and culturability of a strain of Acetobacter pasteurianus and a strain of Brettanomyces bruxellensis isolated from wine. J. Appl. Microbiol. 2005, 98, 862–871. [Google Scholar] [CrossRef]
- Cvetković, D.; Markov, S.; Djurić, M.; Savić, D.; Velićanski, A. Specific interfacial area as a key variable in scaling-up Kombucha fermentation. J. Food Eng. 2008, 85, 387–392. [Google Scholar] [CrossRef]
- Czerny, M.; Christlbauer, M.; Christlbauer, M.; Fischer, A.; Granvogl, M.; Hammer, M.; Hartl, C.; Hernandez, N.M.; Schieberle, P. Re-investigation on odour thresholds of key food aroma compounds and development of an aroma language based on odour qualities of defined aqueous odorant solutions. Eur. Food Res. Technol. 2008, 228, 265–273. [Google Scholar] [CrossRef]
- Meilgaard, M.C. Individual differences in sensory threshold for aroma chemicals added to beer. Food Qual. Prefer. 1993, 4, 153–167. [Google Scholar] [CrossRef]
- Hartwig, P.; McDaniel, M.R. Flavor Characteristics of Lactic, Malic, Citric, and Acetic Acids at Various PH Levels. J. Food Sci. 1995, 60, 384–388. [Google Scholar] [CrossRef]
- Freer, S.; Dien, B.; Matsuda, S. Production of acetic acid by Dekkera/Brettanomyces yeasts under conditions of constant pH. World J. Microbiol. Biotechnol. 2003, 19, 101–105. [Google Scholar] [CrossRef]
- Neta, E.R.D.C.; Johanningsmeier, S.D.; McFeeters, R.F. The Chemistry and Physiology of Sour Taste—A Review. J. Food Sci. 2007, 72, R33–R38. [Google Scholar] [CrossRef]
- Drysdale, G.S.; Fleet, G.H. Acetic Acid Bacteria in Winemaking: A Review. Am. J. Enol. Vitic. 1988, 39, 143–154. [Google Scholar]
- Ho, V.; Zhao, J.; Fleet, G. The effect of lactic acid bacteria on cocoa bean fermentation. Int. J. Food Microbiol. 2015, 205, 54–67. [Google Scholar] [CrossRef]
- Capozzi, V.; Garofalo, C.; Chiriatti, M.A.; Grieco, F.; Spano, G. Microbial terroir and food innovation: The case of yeast biodiversity in wine. Microbiol. Res. 2015, 181, 75–83. [Google Scholar] [CrossRef]
- Styger, G.; Prior, B.; Bauer, F.F. Wine flavor and aroma. J. Ind. Microbiol. Biotechnol. 2011, 38, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.; Baruzzi, F.; Cocolin, L.; Malfeito-Ferreira, M. Emerging technologies to control Brettanomyces spp. in wine: Recent advances and future trends. Trends Food Sci. Technol. 2020, 99, 88–100. [Google Scholar] [CrossRef]
- Serra-Colomer, M.; Funch, B.; Forster, J. The raise of Brettanomyces yeast species for beer production. Curr. Opin. Biotechnol. 2019, 56, 30–35. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa (ASV) | Metabarcoding | Shotgun Sequencing | |
|---|---|---|---|
| Relative Abundance | Prevalence 1 | Proportion of Kmers 2 | |
| Fungi | |||
| g__Brettanomyces | 0.813 ± 0.259 | 0.990 | 0.8884 |
| g__Zygosaccharomyces | 0.068 ± 0.170 | 0.625 | 0.0173 |
| s__Starmerella davenportii | 0.047 ± 0.124 | 0.481 | 0.016 |
| s__Lachancea fermentati | 0.038 ± 0.141 | 0.385 | 0.0086 |
| k__Fungi | 0.012 ± 0.050 | 0.183 | NA |
| f__Saccharomycetales_unidentified | 0.008 ± 0.072 | 0.019 | NA |
| g__Candida | 0.007 ± 0.062 | 0.019 | 0.0005 |
| g__Hanseniaspora | 0.004 ± 0.021 | 0.125 | 0.0038 |
| g__Pichia | 0.001 ± 0.011 | 0.010 | 0.0043 |
| p__Ascomycota | 0.001 ± 0.005 | 0.058 | NA |
| g__Kregervanrija | 0.001 ± 0.004 | 0.019 | 0.0076 |
| Bacteria | |||
| g__Komagataeibacter | 0.809 ± 0.299 | 0.971 | 0.7088 |
| Lactobacillales total | 0.129 | 0.0589 | |
| f__Lactobacillaceae | 0.072 ± 0.167 | 0.394 | NA |
| o__Lactobacillales_unidentified | 0.050 ± 0.117 | 0.327 | NA |
| g__Oenococcus | 0.007 ± 0.061 | 0.077 | 0.019 |
| g__Tanticharoenia | 0.039 ± 0.130 | 0.462 | 0.0003 |
| g__Acetobacter | 0.012 ± 0.055 | 0.173 | 0.117 |
| f__Acetobacteraceae | 0.006 ± 0.030 | 0.144 | NA |
| f__Bacillaceae_unidentified | 0.005 ± 0.040 | 0.029 | 0.0112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, K.; Curtin, C. Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America. Microorganisms 2021, 9, 1060. https://doi.org/10.3390/microorganisms9051060
Harrison K, Curtin C. Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America. Microorganisms. 2021; 9(5):1060. https://doi.org/10.3390/microorganisms9051060
Chicago/Turabian StyleHarrison, Keisha, and Chris Curtin. 2021. "Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America" Microorganisms 9, no. 5: 1060. https://doi.org/10.3390/microorganisms9051060
APA StyleHarrison, K., & Curtin, C. (2021). Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America. Microorganisms, 9(5), 1060. https://doi.org/10.3390/microorganisms9051060