
Use of an Alignment-Free Method for the Geographical Discrimination of GTPVs Based on the GPCR Sequences

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequencing of New Isolates
2.2. Collection of GTPVs for Alignment-Free Method
2.3. Distances Calculation and Visualization
3. Results
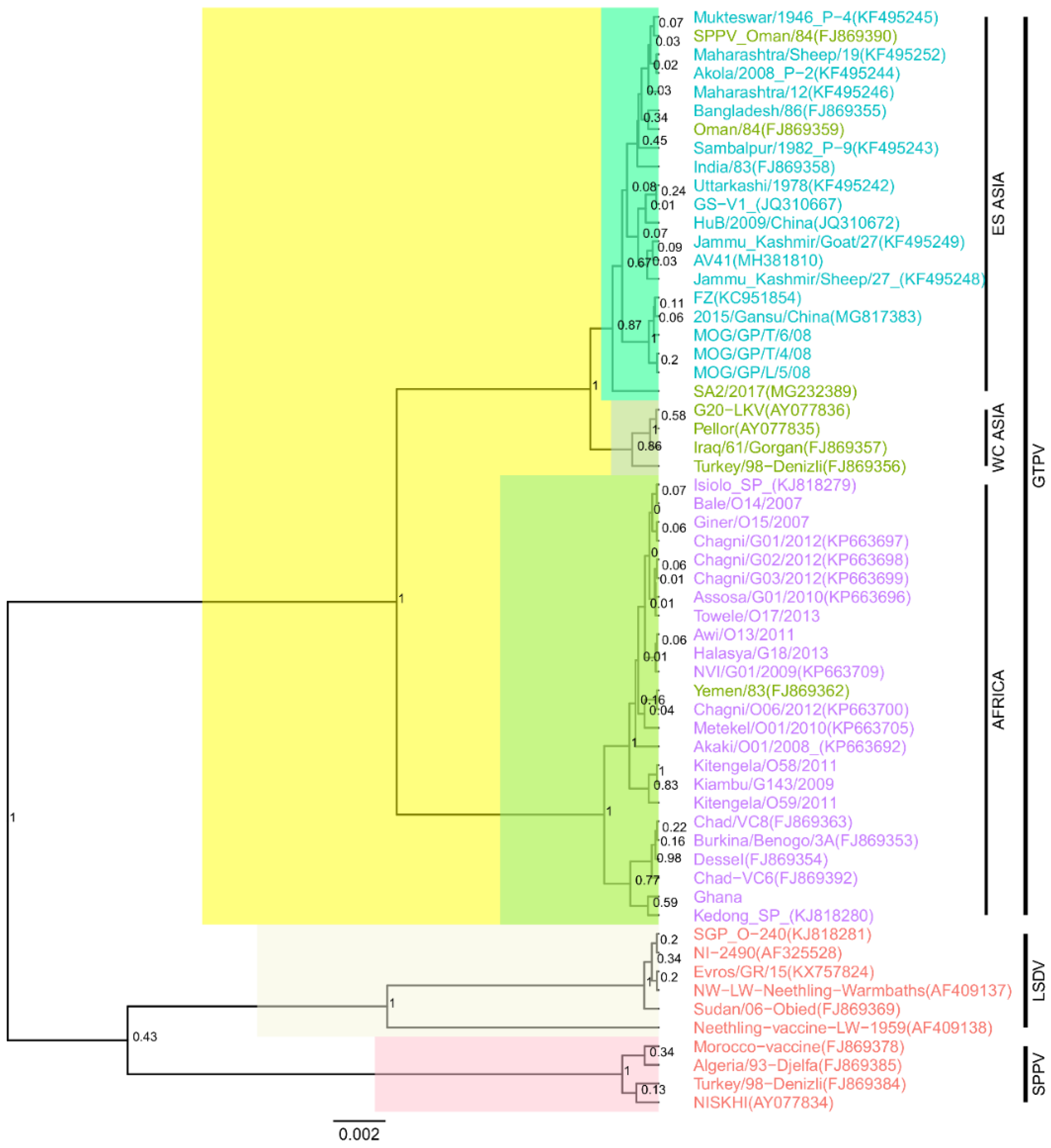
3.1. Sequencing and Phylogenetic Reconstructions
3.2. Distances Calculation and Visualization Using Alignment-Free Methods
3.3. Amino Acids Profiles of GTPVs’ GPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buller, R.M.; Arif, B.M.; Black, D.N.; Dumbell, K.R.; Esposito, J.J.; Lefkowitz, E.J.; McFadden, G.; Moss, B.; Mercer, A.A.; Moyer, R.W.; et al. Family poxviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses. Eighth Report of the International Committee on Taxonomy of Viruses; Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2005; pp. 117–133. [Google Scholar]
- Le Goff, C.; Lamien, C.E.; Fakhfakh, E.; Chadeyras, A.; Aba-Adulugba, E.; Libeau, G.; Tuppurainen, E.; Wallace, D.B.; Adam, T.; Silber, R.; et al. Capripoxvirus G-protein-coupled chemokine receptor: A host-range gene suitable for virus animal origin discrimination. J. Gen. Virol. 2009, 90, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.K.; Roychoudhury, P.; Kawlni, L.; Lalmuanpuia, J.; Dey, A.; Muthuchelvan, D.; Mandakini, R.; Sarkar, A.; Ramakrishnan, M.A.; Subudhi, P.K. An outbreak of Goatpox virus infection in Wild Red Serow (Capricornis rubidus) in Mizoram, India. Transbound. Emerg. Dis. 2019, 66, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Kitching, R.P.; Taylor, W.P. Clinical and antigenic relationship between isolates of sheep and goat pox viruses. Trop. Anim. Health Prod. 1985, 17, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Lamien, C.E.; Lelenta, M.; Goger, W.; Silber, R.; Tuppurainen, E.; Matijevic, M.; Luckins, A.G.; Diallo, A. Real time PCR method for simultaneous detection, quantitation and differentiation of capripoxviruses. J. Virol. Methods 2011, 171, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Lamien, C.E.; Le Goff, C.; Silber, R.; Wallace, D.B.; Gulyaz, V.; Tuppurainen, E.; Madani, H.; Caufour, P.; Adam, T.; El Harrak, M.; et al. Use of the Capripoxvirus homologue of Vaccinia virus 30 kDa RNA polymerase subunit (RPO30) gene as a novel diagnostic and genotyping target: Development of a classical PCR method to differentiate Goat poxvirus from Sheep poxvirus. Vet. Microbiol. 2011, 149, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Venter, E.H.; Shisler, J.L.; Gari, G.; Mekonnen, G.A.; Juleff, N.; Lyons, N.A.; De Clercq, K.; Upton, C.; Bowden, T.R.; et al. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transbound. Emerg. Dis. 2017, 64, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Gelaye, E.; Belay, A.; Ayelet, G.; Jenberie, S.; Yami, M.; Loitsch, A.; Tuppurainen, E.; Grabherr, R.; Diallo, A.; Lamien, C.E. Capripox disease in Ethiopia: Genetic differences between field isolates and vaccine strain, and implications for vaccination failure. Antivir. Res. 2015, 119, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Santhamani, R.; Venkatesan, G.; Minhas, S.K.; Shivachandra, S.B.; Muthuchelvan, D.; Pandey, A.B.; Ramakrishnan, M.A. Detection and characterization of atypical capripoxviruses among small ruminants in India. Virus Genes 2015, 51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Santhamani, R.; Yogisharadhya, R.; Venkatesan, G.; Shivachandra, S.B.; Pandey, A.B.; Ramakrishnan, M.A. Molecular characterization of Indian sheeppox and goatpox viruses based on RPO30 and GPCR genes. Virus Genes 2014, 49, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Jia, H.; Chen, G.; He, X.; Fang, Y.; Wang, X.; Guan, Q.; Zeng, S.; Cui, Q.; Jing, Z. Phylogenetic analysis of Chinese sheeppox and goatpox virus isolates. Virol. J. 2012, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vinga, S.; Almeida, J. Alignment-free sequence comparison—A review. Bioinformatics 2003, 19, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Bonham-Carter, O.; Steele, J.; Bastola, D. Alignment-free genetic sequence comparisons: A review of recent approaches by word analysis. Brief. Bioinform. 2013, 15, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Kitching, R.P.; McGrane, J.J.; Taylor, W.P. Capripox in the Yemen Arab Republic and the Sultanate of Oman. Trop. Anim. Health Prod. 1986, 18, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Knowles, N.J.; Paton, D.J. Combining livestock trade patterns with phylogenetics to help understand the spread of foot and mouth disease in sub-Saharan Africa, the Middle East and Southeast Asia. Rev. Sci. Tech. 2011, 30, 63. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Isolate | Year of Collection | Species | Country | Region | Accession Number |
|---|---|---|---|---|---|
| * Awi/O13/2011 | 2011 | Sheep | Ethiopia | Africa | MN161836 |
| * Bale/O14/2007 | 2007 | Sheep | Ethiopia | Africa | MN161837 |
| * Giner/O15/2007 | 2007 | Goat | Ethiopia | Africa | MN161838 |
| * Towele/O17/2013 | 2013 | Goat | Ethiopia | Africa | MN161839 |
| * Halasya/G18/2013 | 2013 | Goat | Ethiopia | Africa | MN161840 |
| * Kitengela/O58/2011 | 2011 | Sheep | Kenya | Africa | MN161841 |
| * Kitengela/O59/2011 | 2011 | Goat | Kenya | Africa | MN161842 |
| * Kiambu/G143/2009 | 2009 | Goat | Kenya | Africa | MN161843 |
| * Ghana | Unknown | Goat | Ghana | Africa | MN161844 |
| Assosa/G01/2010 | 2010 | Goat | Ethiopia | Africa | KP663696 |
| Chagni/O06/2012 | 2012 | Sheep | Ethiopia | Africa | KP663700 |
| Chagni/G03/2012 | 2012 | Goat | Ethiopia | Africa | KP663699 |
| Chagni/G02/2012 | 2012 | Goat | Ethiopia | Africa | KP663698 |
| Chagni/G01/2012 | 2012 | Goat | Ethiopia | Africa | KP663697 |
| NVI/G01/2009) | 2009 | Goat | Ethiopia | Africa | KP663709 |
| Yemen/83 | 1983 | Goat | Yemen | West Asia | FJ869362 |
| Metekel/O01/2010 | 2010 | Sheep | Ethiopia | Africa | KP663705 |
| Akaki/O01/2008 | 2008 | Sheep | Ethiopia | Africa | KP663692 |
| Isiolo_SP | 1959 | Sheep | Kenya | Africa | KJ818279 |
| Chad-VC6 | Unknown | Goat | Chad | Africa | FJ869392 |
| Burkina/Benogo/3A | Unknown | Goat | Burkina Faso | Africa | FJ869353 |
| DesseI | Unknown | Goat | Unknown | Africa | FJ869354 |
| Chad/VC8 | Unknown | Goat | Chad | Africa | FJ869363 |
| Kedong_SP | 1955 | Sheep | Kenya | Africa | KJ818280 |
| G20-LKV | 2000 | Goat | Kazakhstan | Central Asia | AY077836 |
| SA2/2017 | 2017 | Goat | Saudi Arabia | West Asia | MG232389 |
| Iraq/61/Gorgan | 1961 | Goat | Iraq | West Asia | FJ869357 |
| Pellor | 2000 | Goat | Kazakhstan | Central Asia | AY077835 |
| Turkey/98-Denizli | 1998 | Goat | Turkey | West Asia | FJ869356 |
| Maharashtra/12 | 2008 | Goat | India | South Asia | KF495246 |
| Maharashtra/Sheep/19 | 2010 | Sheep | India | South Asia | KF495252 |
| Mukteswar/1946_P-4 | 1946 | Goat | India | South Asia | KF495245 |
| * MOG/GP/L/5/08 | 2008 | Goat | Mongolia | East Asia | MN161845 |
| * MOG/GP/T/4/08 | 2008 | Goat | Mongolia | East Asia | MN161846 |
| * MOG/GP/T/6/08 | 2008 | Goat | Mongolia | East Asia | MN161847 |
| Akola/2008_P-2 | 2008 | Goat | India | South Asia | KF495244 |
| Sambalpur/1982_P-9 | 1982 | Goat | India | South Asia | KF495243 |
| Uttarkashi/1978 | 1978 | Goat | India | South Asia | KF495242 |
| Jammu_Kashmir/Goat/27 | 2013 | Goat | India | South Asia | KF495249 |
| Jammu_Kashmir/Sheep/27 | 2013 | Sheep | India | South Asia | KF495248 |
| GS-V1 | 2011 | Unknown | China | East Asia | JQ310667 |
| India/83 | 1983 | Goat | India | South Asia | FJ869358 |
| SPPV_Oman/84 | 1984 | Sheep | Oman | West Asia | FJ869390 |
| Oman/84 | 1984 | Goat | Oman | West Asia | FJ869359 |
| Bangladesh/86 | 1986 | Goat | Bangladesh | South Asia | FJ869355 |
| FZ | 2012 | Goat | China | East Asia | KC951854 |
| AV41 | 2018 | Goat | China | East Asia | MH381810 |
| 2015/Gansu/China | 2015 | Unknown | China | East Asia | MG817383 |
| HuB/2009/China | 2009 | Goat | China | East Asia | JQ310672 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chibssa, T.R.; Liu, Y.; Sombo, M.; Lichoti, J.K.; Erdenebaatar, J.; Boldbaatar, B.; Grabherr, R.; Settypalli, T.B.K.; Berguido, F.J.; Loitsch, A.; et al. Use of an Alignment-Free Method for the Geographical Discrimination of GTPVs Based on the GPCR Sequences. Microorganisms 2021, 9, 855. https://doi.org/10.3390/microorganisms9040855
Chibssa TR, Liu Y, Sombo M, Lichoti JK, Erdenebaatar J, Boldbaatar B, Grabherr R, Settypalli TBK, Berguido FJ, Loitsch A, et al. Use of an Alignment-Free Method for the Geographical Discrimination of GTPVs Based on the GPCR Sequences. Microorganisms. 2021; 9(4):855. https://doi.org/10.3390/microorganisms9040855
Chicago/Turabian StyleChibssa, Tesfaye Rufael, Yang Liu, Melaku Sombo, Jacqueline Kasiiti Lichoti, Janchivdorj Erdenebaatar, Bazartseren Boldbaatar, Reingard Grabherr, Tirumala Bharani K. Settypalli, Francisco J. Berguido, Angelika Loitsch, and et al. 2021. "Use of an Alignment-Free Method for the Geographical Discrimination of GTPVs Based on the GPCR Sequences" Microorganisms 9, no. 4: 855. https://doi.org/10.3390/microorganisms9040855
APA StyleChibssa, T. R., Liu, Y., Sombo, M., Lichoti, J. K., Erdenebaatar, J., Boldbaatar, B., Grabherr, R., Settypalli, T. B. K., Berguido, F. J., Loitsch, A., Damena, D., Cattoli, G., Diallo, A., & Lamien, C. E. (2021). Use of an Alignment-Free Method for the Geographical Discrimination of GTPVs Based on the GPCR Sequences. Microorganisms, 9(4), 855. https://doi.org/10.3390/microorganisms9040855