
Distribution of Prophages in the Oenococcus oeni Species

 , ,
, ,
Abstract
1. Introduction
2. Material and Methods
2.1. Data Collection and Identification of Candidate Prophage Sequences in Complete Genomes of O. oeni
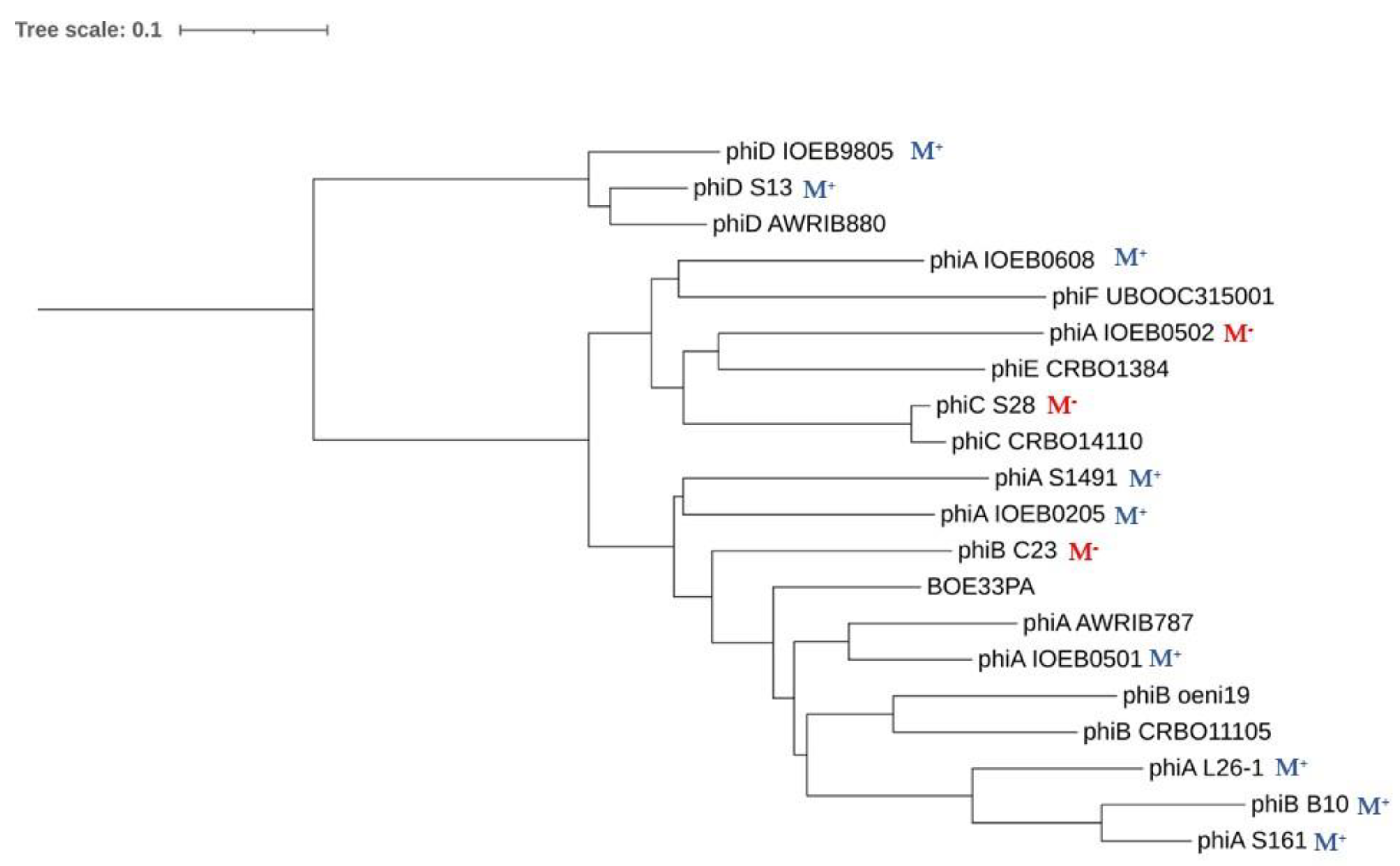
2.2. Comparisons and Phylogenetic Analyses
2.3. PCR
2.4. Induction with MC
3. Results and Discussion
3.1. The Genome of O. oeni Is Replete with Putative Prophages
3.2. Phage Remnants Also Use tRNA Sites in O. oeni
3.3. Chromosomal Location and Genomic Context of Prophages and PR
3.4. Lysogeny Is Widespread among Wine Strains from Phylogroup A
3.5. Integrase Phylogeny
3.6. Correlation of Phage Phylogeny with attB Location
3.6.1. SSR of IntF and IntB Prophages
3.6.2. SRR of PRF Elements
3.7. Most attP Regions in Oenophages Derive from Two Distinct Sequences
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Grandvalet, C. Oenococcus oeni: Queen of the cellar, nightmare of geneticists. Microbiology 2017, 163, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, M.P.G.; Lucas, P.M. Distribution of Oenococcus oeni populations in natural habitats. Appl. Microbiol. Biotechnol. 2019, 103, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Mills, D.A.; Rawsthorne, H.; Parker, C.; Tamir, D.; Makarova, K. Genomic analysis of Oenococcus oeni PSU-1 and its relevance to winemaking. FEMS Microbiol. Rev. 2005, 29, 465–475. [Google Scholar] [CrossRef]
- Borneman, A.R.; McCarthy, J.M.; Chambers, P.J.; Bartowsky, E.J. Comparative analysis of the Oenococcus oeni pan genome reveals genetic diversity in industrially-relevant pathways. BMC Genom. 2012, 13, 373. [Google Scholar] [CrossRef]
- Dimopoulou, M.; Vuillemin, M.; Campbell-Sills, H.; Lucas, P.M.; Ballestra, P.; Miot-Sertier, C.; Favier, M.; Coulon, J.; Moine, V.; Doco, T.; et al. Exopolysaccharide (EPS) synthesis by Oenococcus oeni: From genes to phenotypes. PLoS ONE 2014, 9, e98898. [Google Scholar] [CrossRef]
- Sternes, P.R.; Borneman, A.R. Consensus Pan-Genome Assembly of the Specialised Wine Bacterium Oenococcus oeni. BMC Genom. 2016, 17, 813. [Google Scholar]
- Campbell-Sills, H.; Khoury, M.E.; Favier, M.; Romano, A.; Biasioli, F.; Spano, G.; Sherman, D.J.; Bouchez, O.; Coton, E.; Coton, M.; et al. Phylogenomic analysis of Oenococcus oeni reveals specific domestication of strains to cider and wines. Genome Biol. Evol. 2015, 7, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, M.; Campbell-Sills, H.; Salin, F.; Guichoux, E.; Claisse, O.; Lucas, P.M. Biogeography of Oenococcus oeni Reveals Distinctive but Nonspecific Populations in Wine-Producing Regions. Appl. Environ. Microbiol. 2017, 83, e02322-16. [Google Scholar] [CrossRef]
- Lorentzen, M.P.; Campbell-Sills, H.; Jorgensen, T.S.; Nielsen, T.K.; Coton, M.; Coton, E.; Hansen, L.; Lucas, P.M. Expanding the biodiversity of Oenococcus oeni through comparative genomics of apple cider and kombucha strains. BMC Genom. 2019, 20, 330. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Sills, H.; El Khoury, M.; Gammacurta, M.; Miot-Sertier, C.; Dutilh, L.; Vestner, J.; Capozzi, V.; Sherman, D.; Hubert, C.; Claisse, O.; et al. Two different Oenococcus oeni lineages are associated to either red or white wines in Burgundy: Genomics and metabolomics insights. OENO One 2017, 51, 309. [Google Scholar] [CrossRef]
- Marcobal, A.M.; Sela, D.A.; Wolf, Y.I.; Makarova, K.S.; Mills, D.A. Role of hypermutability in the evolution of the genus Oenococcus. J. Bacteriol. 2008, 190, 564–570. [Google Scholar] [CrossRef]
- Meier, P.; Wackernagel, W. Impact of mutS inactivation on foreign DNA acquisition by natural transformation in Pseudomonas stutzeri. J. Bacteriol. 2005, 187, 143–154. [Google Scholar] [CrossRef]
- El Gharniti, F.; Dols-Lafargue, M.; Bon, E.; Claisse, O.; Miot-Sertier, C.; Lonvaud, A.; Le Marrec, C. IS30 elements are mediators of genetic diversity in Oenococcus oeni. Int. J. Food Microbiol. 2012, 158, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Favier, M.; Bilhère, E.; Lonvaud-Funel, A.; Moine, V.; Lucas, P.M. Identification of pOENI-1 and related plasmids in Oenococcus oeni strains performing the malolactic fermentation in wine. PLoS ONE 2012, 7, e49082. [Google Scholar] [CrossRef] [PubMed]
- Bon, E.; Delaherche, A.; Bilhère, E.; De Daruvar, A.; Lonvaud-Funel, A.; Le Marrec, C. Oenococcus oeni genome plasticity is associated with fitness. Appl. Environ. Microbiol. 2009, 75, 2079–2090. [Google Scholar] [CrossRef]
- Poblet-Icart, M.; Bordons, A.; Lonvaud-Funel, A. Lysogeny of Oenococcus oeni (syn. Leuconostoc oenos) and study of their induced bacteriophages. Curr. Microbiol. 1998, 36, 365–369. [Google Scholar] [CrossRef]
- Jaomanjaka, F.; Ballestra, P.; Dols-Lafargue, M.; Le Marrec, C. Expanding the diversity of oenococcal bacteriophages: Insights into a novel group based on the integrase sequence. Int. J. Food Microbiol. 2013, 166, 331–340. [Google Scholar] [CrossRef]
- Philippe, C.; Jaomanjaka, F.; Claisse, O.; Laforgue, R.; Maupeu, J.; Petrel, M.; Le Marrec, C. A survey of oenophages during wine making reveals a novel group with unusual genomic characteristics. Int. J. Food Microbiol. 2017, 257, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef]
- Wahl, A.; Battesti, A.; Ansaldi, M. Prophages in Salmonella enterica: A driving force in reshaping the genome and physiology of their bacterial host? Mol. Microbiol. 2019, 111, 303–316. [Google Scholar] [CrossRef]
- Ruiz-Cruz, S.; Parlindungan, E.; Erazo Garzon, A.; Alqarni, M.; Lugli, G.A.; Ventura, M.; van Sinderen, D.; Mahony, J. Lysogenization of a Lactococcal Host with Three Distinct Temperate Phages Provides Homologous and Heterologous Phage Resistance. Microorganisms 2020, 8, 1685. [Google Scholar] [CrossRef]
- Matos, R.C.; Lapaque, N.; Rigottier-Gois, L.; Debarbieux, L.; Meylheuc, T.; Gonzalez-Zorn, B.; Repoila, F.; Lopes, M.d.F.; Serror, P. Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLoS Genet. 2013, 9, e1003539. [Google Scholar] [CrossRef]
- Aucouturier, A.; Chain, F.; Langella, P.; Bidnenko, E. Characterization of a Prophage-Free Derivative Strain of Lactococcus lactis ssp. lactis IL1403 Reveals the Importance of Prophages for Phenotypic Plasticity of the Host. Front. Microbiol. 2018, 9, 2032. [Google Scholar] [CrossRef]
- Lynn-Bell, N.L.; Strand, M.R.; Oliver, K.M. Bacteriophage acquisition restores protective mutualism. Microbiology 2019, 165, 985–989. [Google Scholar] [CrossRef] [PubMed]
- De Sordi, L.; Lourenço, M.; Debarbieux, L. “I will survive”: A tale of bacteriophage-bacteria coevolution in the gut. Gut Microbes 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Braga, L.P.P.; Soucy, S.M.; Amgarten, D.E.; da Silva, A.M.; Setubal, J.C. Bacterial Diversification in the light of the interactions with phages: The genetic symbionts and their role in ecological speciation. Front. Ecol. Evol. 2018, 6, 6. [Google Scholar] [CrossRef]
- Szafrański, S.P.; Kilian, M.; Yang, I.; Der Wieden, G.B.; Winkel, A.; Hegermann, J.; Stiesch, M. Diversity patterns of bacteriophages infecting Aggregatibacter and Haemophilus species across clades and niches. ISME J. 2019, 13, 2500–2522. [Google Scholar] [CrossRef] [PubMed]
- Badotti, F.; Moreira, A.P.B.; Tonon, L.A.C.; De Lucena, B.T.L.; Gomes, F.D.C.O.; Krüger, R.; Thompson, C.C.; De Morais, M.A.; Rosa, C.A.; Thompson, F.L. Oenococcus alcoholitolerans sp. nov., a lactic acid bacteria isolated from cachaça and ethanol fermentation processes. Antonie Van Leeuwenhoek 2004, 106, 1259–1267. [Google Scholar] [CrossRef]
- Endo, A.; Okada, S. Oenococcus kitaharae sp. nov., a non-acidophilic and non-malolactic-fermenting Oenococcus isolated from a composting distilled shochu residue. Int. J. Syst. Evol. Microbiol. 2006, 56, 2345–2348. [Google Scholar] [CrossRef]
- Cousin, F.J.; Le Guellec, R.; Chagnot, C.; Goux, D.; Dalmasso, M.; Laplace, J.M.; Cretenet, M. Oenococcus sicerae sp. nov., isolated from French cider. Syst. Appl. Microbiol. 2019, 42, 302–308. [Google Scholar] [CrossRef]
- Verce, M.; De Vuyst, L.; Weckx, S. The metagenome-assembled genome of Candidatus Oenococcus aquikefiri from water kefir represents the species Oenococcus sicerae. Food Microbiol. 2020, 88, 103402. [Google Scholar] [CrossRef]
- McNair, K.; Aziz, R.K.; Pusch, G.D.; Overbeek, R.; Dutilh, B.E.; Edwards, R. Phage Genome Annotation Using the RAST Pipeline. Methods Mol. Biol. 2018, 1681, 231–238. [Google Scholar]
- Czajkowski, R. May the Phage be With You? Prophage-Like Elements in the Genomes of Soft Rot Pectobacteriaceae: Pectobacterium spp. and Dickeya spp. Front. Microbiol. 2019, 10, 138. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Philippe, C.; Krupovic, M.; Jaomanjaka, F.; Claisse, O.; Petrel, M.; Le Marrec, C. Bacteriophage GC1, a novel Tectivirus Infecting Gluconobacter cerinus, an acetic acid bacterium associated with wine-making. Viruses 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- São-José, C.; Santos, S.; Nascimento, J.; Brito-Madurro, A.G.; Parreira, R.; Santos, M.A. Diversity in the lysis-integration region of oenophage genomes and evidence for multiple tRNA loci, as targets for prophage integration in Oenococcus oeni. Virology 2004, 325, 82–95. [Google Scholar] [CrossRef]
- Petersen, A.; Josephsen, J.; Johnsen, M.G. TPW22, a lactococcal temperate phage with a site-specific integrase closely related to Streptococcus thermophilus phage integrases. J. Bacteriol. 1999, 181, 7034–7042. [Google Scholar] [CrossRef]
- Van der Ploeg, J.R. Characterization of Streptococcus gordonii Prophage PH15: Complete Genome Sequence and Functional Analysis of Phage-Encoded Integrase and Endolysin. Microbiology 2008, 154, 2970–2978. [Google Scholar] [CrossRef]
- Bobay, L.M.; Rocha, P.C.; Touchon, M. The Adaptation of Temperate Bacteriophages to Their Host Genomes. Mol. Biol. Evol. 2013, 30, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Rocha, E.P. Coevolution of the Organization and Structure of Prokaryotic Genomes. Cold Spring Harb. Perspect. Biol. 2016, 8, a018168. [Google Scholar] [CrossRef]
- Kopejtka, K.; Lin, Y.; Jakubovičová, M.; Koblížek, M.; Tomasch, J. Clustered Core- and Pan-Genome Content on Rhodobacteraceae Chromosomes. Genome Biol. Evol. 2019, 11, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.N.; Mettert, E.L.; Fishman-Engel, D.R.; Roggiani, M.; Kiley, P.J.; Goulian, M. Phage integration alters the respiratory strategy of its host. eLife 2019, 8, e49081. [Google Scholar] [CrossRef]
- Takada, H.; Yoshikawa, H. Essentiality and function of WalK/WalR two-component system: The past, present, and future of research. Biosci. Biotechnol. Biochem. 2018, 82, 741–751. [Google Scholar] [CrossRef]
- Li, C.; Sun, J.W.; Zhang, G.F.; Liu, L.B. Effect of the absence of the CcpA gene on growth, metabolic production, and stress tolerance in Lactobacillus delbrueckii ssp. bulgaricus. J. Dairy Sci. 2016, 99, 104–111. [Google Scholar] [CrossRef]
- Knowles, B.; Silveira, C.B.; Bailey, B.A.; Barott, K.; Cantu, V.A.; Cobián-Güemes, A.G.; Coutinho, F.H.; Dinsdale, E.A.; Felts, B.; Furby, K.A.; et al. Lytic to temperate switching of viral communities. Nature 2016, 531, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.A.P.; Harris, H.M.B.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Le Bourgeois, P.; Bugarel, M.; Campo, N.; Daveran-Mingot, M.L.; Labonté, J.; Lanfranchi, D.; Lautier, T.; Pagès, C.; Ritzenthaler, P. The unconventional Xer recombination machinery of Streptococci/Lactococci. PLoS Genet. 2007, 3, e117. [Google Scholar] [CrossRef]
- Zúñiga, M.; Pardo, I.; Ferrer, S. Transposons Tn916 and Tn925 can transfer from Enterococcus faecalis to Leuconostoc oenos. FEMS Microbiol. Lett. 1996, 135, 179–185. [Google Scholar] [CrossRef]
- Mesas, J.M.; Rodriguez, M.C.; Alegre, M.T. Characterization of lactic acid bacteria from musts and wines of three consecutive vintages of Ribeira Sacra. Lett. Appl. Microbiol. 2011, 52, 258–268. [Google Scholar] [CrossRef]
- Groth, A.C.; Calos, M.P. Phage integrases: Biology and applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef]
- Jaomanjaka, F.; Claisse, O.; Blanche-Barbat, M.; Petrel, M.; Ballestra, P.; Le Marrec, C. Characterization of a new virulent phage infecting the lactic acid bacterium Oenococcus oeni. Food Microbiol. 2016, 54, 167–177. [Google Scholar] [CrossRef]
- Auvray, F.; Coddeville, M.; Ordonez, R.C.; Ritzenthaler, P. Unusual structure of the attB site of the site-specific recombination system of Lactobacillus delbrueckii bacteriophage mv4. J. Bacteriol. 1999, 181, 7385–7389. [Google Scholar] [CrossRef]
- Campbell, A. Phage integration and chromosome structure. A personal history. Annu. Rev. Genet. 2007, 41, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Landy, A.; Ross, W. Viral integration and excision: Structure of the lambda att sites. Science 1977, 197, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Wojciak, J.M.; Sarkar, D.; Landy, A.; Clubb, R.T. Arm-site binding by lambda -integrase: Solution structure and functional characterization of its amino-terminal domain. Proc. Natl. Acad. Sci. USA 2002, 99, 3434–3439. [Google Scholar] [CrossRef]
- Cho, E.H.; Gumport, R.I.; Gardner, J.F. Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. J. Bacteriol. 2002, 184, 5200–5203. [Google Scholar] [CrossRef]
- Suzuki, S.; Yoshikawa, M.; Imamura, D.; Abe, K.; Eichenberger, P.; Sato, T. Compatibility of Site-Specific Recombination Units between Mobile Genetic Elements. iScience 2020, 23, 100805. [Google Scholar] [CrossRef] [PubMed]
- Rutkai, E.; György, A.; Dorgai, L.; Weisberg, R.A. Role of Secondary Attachment Sites in Changing the Specificity of Site-Specific Recombination. J. Bacteriol. 2006, 188, 3409–3411. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Kurokawa, K.; Yamashita, A.; Nakata, M.; Tomiyasu, Y.; Okahashi, N.; Kawabata, S.; Yamazaki, K.; Shiba, T.; Yasunaga, T.; et al. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 2003, 13, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Chaïb, A.; Jaomanjaka, F.; Claisse, O.; Lucas, P.M.; Samot, J.; Cambillau, C.; Le Marrec, C. Characterization of the First Virulent Phage Infecting Oenococcus oeni, the Queen of the Cellars. Front. Microbiol. 2021, 11, 596541. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distribution and Type of Prophages | Phylogroups in the O. oeni Species | |||
|---|---|---|---|---|
| A | B | C | D | |
| Number of strains analyzed | 174 | 31 | 21 | 5 |
| Number of mono- and poly-lysogens | 105 | 17 | 8 | 4 |
| Lysogeny (%) | 60 | 55 | 38 | 80 |
| Mono-lysogens (n = 86) and prophage types | ||||
| IntA | 22 | 2 | 0 | 0 |
| IntB | 14 | 1 | 1 | 0 |
| IntC | 20 | 0 | 0 | 0 |
| IntD | 3 | 12 | 4 | 0 |
| IntE | 0 | 0 | 3 | 0 |
| IntF | 0 | 0 | 0 | 4 |
| Poly-lysogens (n = 48) and prophage combinations | ||||
| IntAB | 3 | 1 | 0 | 0 |
| IntAC | 17 | 0 | 0 | 0 |
| IntAD | 8 | 0 | 0 | 0 |
| IntBC | 14 | 0 | 0 | 0 |
| IntABC | 1 | 1 | 0 | 0 |
| IntACD | 3 | 0 | 0 | 0 |
| Phage remnants (PRs) in strains | ||||
| PRC | 174 | 0 | 9 | 0 |
| PRF | 0 | 1 | 1 | 4 |
| PRG | 0 | 0 | 0 | 4 |
| PRH | 18 | 6 | 12 | 5 |
| PRI | 9 | 14 | 15 | 0 |
| Integrase Types | IntA | IntB | IntC | IntD | IntE |
|---|---|---|---|---|---|
| IntB | 69 a/62 b | ||||
| IntC | 68/60 | 84/86 | |||
| IntD | 74/66 | 68/61 | 68/59 | ||
| IntE | 66/61 | 72/69 | 72/68 | 66/60 | |
| IntF | 70/61 | 88/92 | 88/92 | 69/60 | 73/68 |
| Phage-Related Elements | Host Strain and attB Site | Putative Integrase Binding Sites (DR *) on attP | |||||
|---|---|---|---|---|---|---|---|
| P/PR | Strain Harboring the Prophage or PR Name | Int | Name, Phylogroup and Niche | attB Site (bp) | nb | Right/Left Arms | DR Sequences |
| P | IOEB0608 | IntA | IOEB0608 (A/wine) | attBA (tRNAGlu, 16 bp) | 5 | 2/3 | -TTT-GCCACA (x2) -TTT-GCCACA (x2) -TTT-GCCACA (x1) |
| 10MC | IntB-B | IOEB B10 (A/wine) | attBB (tRNALeu, 15 bp) | 5 | 2/3 | --ATCG-CACAA (x5) --ATCG-CACAA (x1) | |
| LAB2013 | IntB-F | LAB2013 (A/wine) | attBF (tRNALeu, 63 bp) | 5 | 2/3 | --AT-G-CACAA (x1) --ATCG-CACAA (x3) --ATCG-CACAA (x1) | |
| IOEBS28 | IntC | IOEB S28 | attBC (tRNALys, 78 bp) | 4 | 2/2 | --AT-G-CACAACAA (x3) --CT-G-CACAACAA (x1) | |
| IOEB9805 | IntD | IOEB9805 (B/wine) | attBD (tRNALeu, 128 bp) | 5 | 2/3 | -TTT-G-CACA (x5) | |
| CRBO1384 | IntE | CRBO1384 | attBE (tRNACyst, 11 bp) | 4 | 2/2 | T-GCCAC-CGTT (x4) | |
| UBOCC315005 | IntF | UBOCC315005 (D/kombucha) | attBF (tRNALeu, 63 bp) | 6 | 2/4 | ATTT-G-CACAA (x1) AT-T-G-CACAA (x2) AA-T-G-CACAA (x3) | |
| PR | PRC | IntC | IOEB S28 | attBC (tRNALys, 78 bp) | 4 | 2/2 | --AT-G-CACAACAA(x3) --CT-G-CACAACAA (x1) |
| PRF1 | IntF1 | DIV5-23 (B/cider) | attBF (tRNALeu, 63 bp) | 5 | 3/2 | ---TGG-TACAA (x4) ---TGGCTACAA (x1) | |
| PRF2 | IntF2 | CRB01381 (C/cider) | attBF (tRNALeu, 63 bp) | 6 | 3/3 | ---TGG-TACAA (x4) ---TAGCTACAA (x1) ---TGG-TCAAA (x1) | |
| PRG | IntG | UBOCC315005 | attBG (tRNACyst, 61 bp) | nf | |||
| PRH1 | IntH1 | AWRIB422 | attBH (tRNASer, 22 bp) | nf | |||
| PRH2 | IntH2 | IOEBC52 | attBH (tRNASer, 22 bp) | nf | |||
| PRI1 | IntI1 | IOEB277 | attBI (tRNASer, 19 bp) | nf | |||
| PRI2 | IntI2 | AWRIB576 | attBI (tRNASer, 19 bp) | nf | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claisse, O.; Chaïb, A.; Jaomanjaka, F.; Philippe, C.; Barchi, Y.; Lucas, P.M.; Le Marrec, C. Distribution of Prophages in the Oenococcus oeni Species. Microorganisms 2021, 9, 856. https://doi.org/10.3390/microorganisms9040856
Claisse O, Chaïb A, Jaomanjaka F, Philippe C, Barchi Y, Lucas PM, Le Marrec C. Distribution of Prophages in the Oenococcus oeni Species. Microorganisms. 2021; 9(4):856. https://doi.org/10.3390/microorganisms9040856
Chicago/Turabian StyleClaisse, Olivier, Amel Chaïb, Fety Jaomanjaka, Cécile Philippe, Yasma Barchi, Patrick M. Lucas, and Claire Le Marrec. 2021. "Distribution of Prophages in the Oenococcus oeni Species" Microorganisms 9, no. 4: 856. https://doi.org/10.3390/microorganisms9040856
APA StyleClaisse, O., Chaïb, A., Jaomanjaka, F., Philippe, C., Barchi, Y., Lucas, P. M., & Le Marrec, C. (2021). Distribution of Prophages in the Oenococcus oeni Species. Microorganisms, 9(4), 856. https://doi.org/10.3390/microorganisms9040856