Recent Molecular Evolution of Human Metapneumovirus (HMPV): Subdivision of HMPV A2b Strains

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Specimens and HMPV Isolation
2.2. Full-Genome Analysis of HMPV
2.3. Maximum Likelihood Phylogenies
2.4. Molecular Clock Phylogenies
2.5. Ethics Statement
3. Results
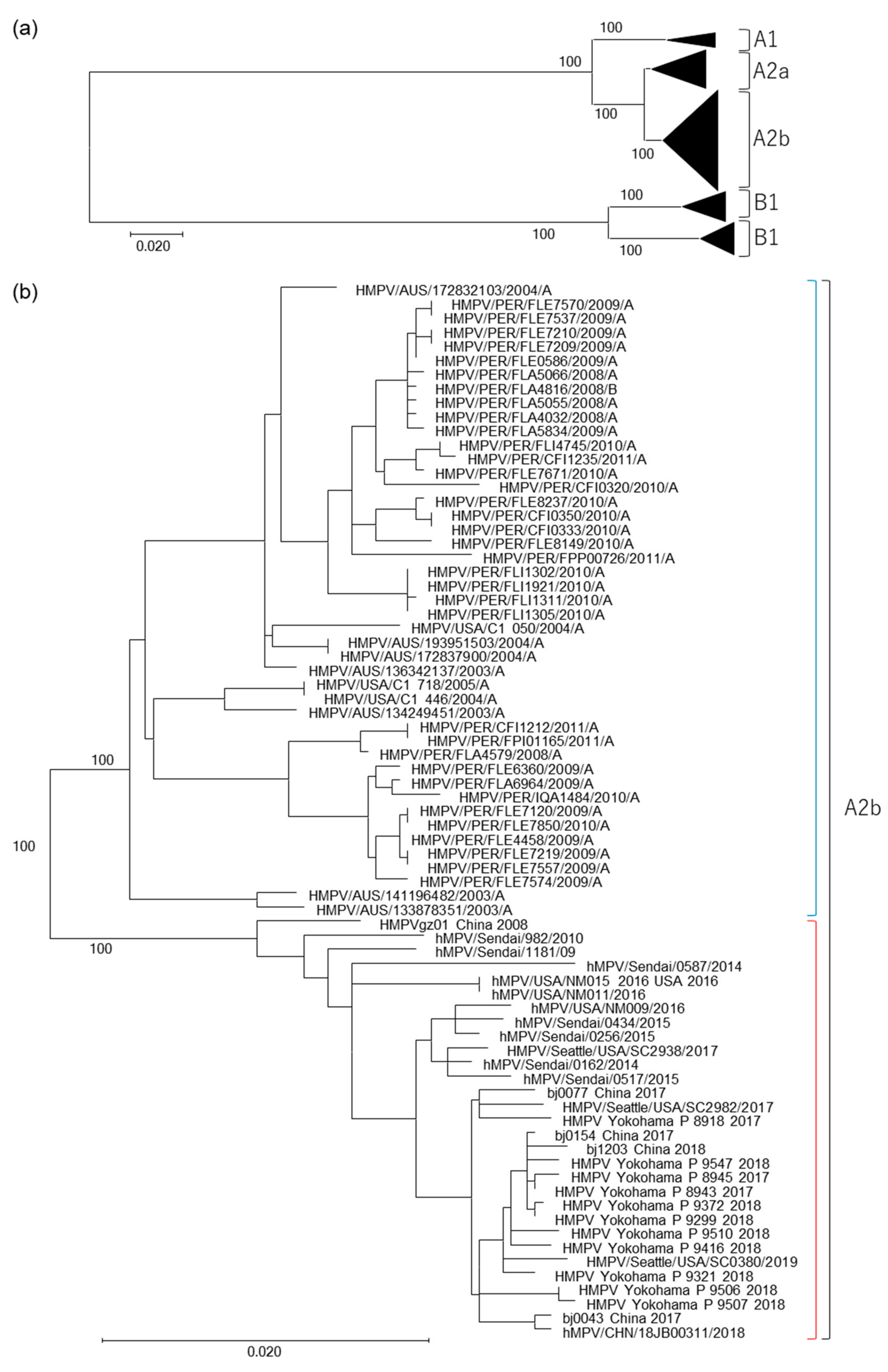
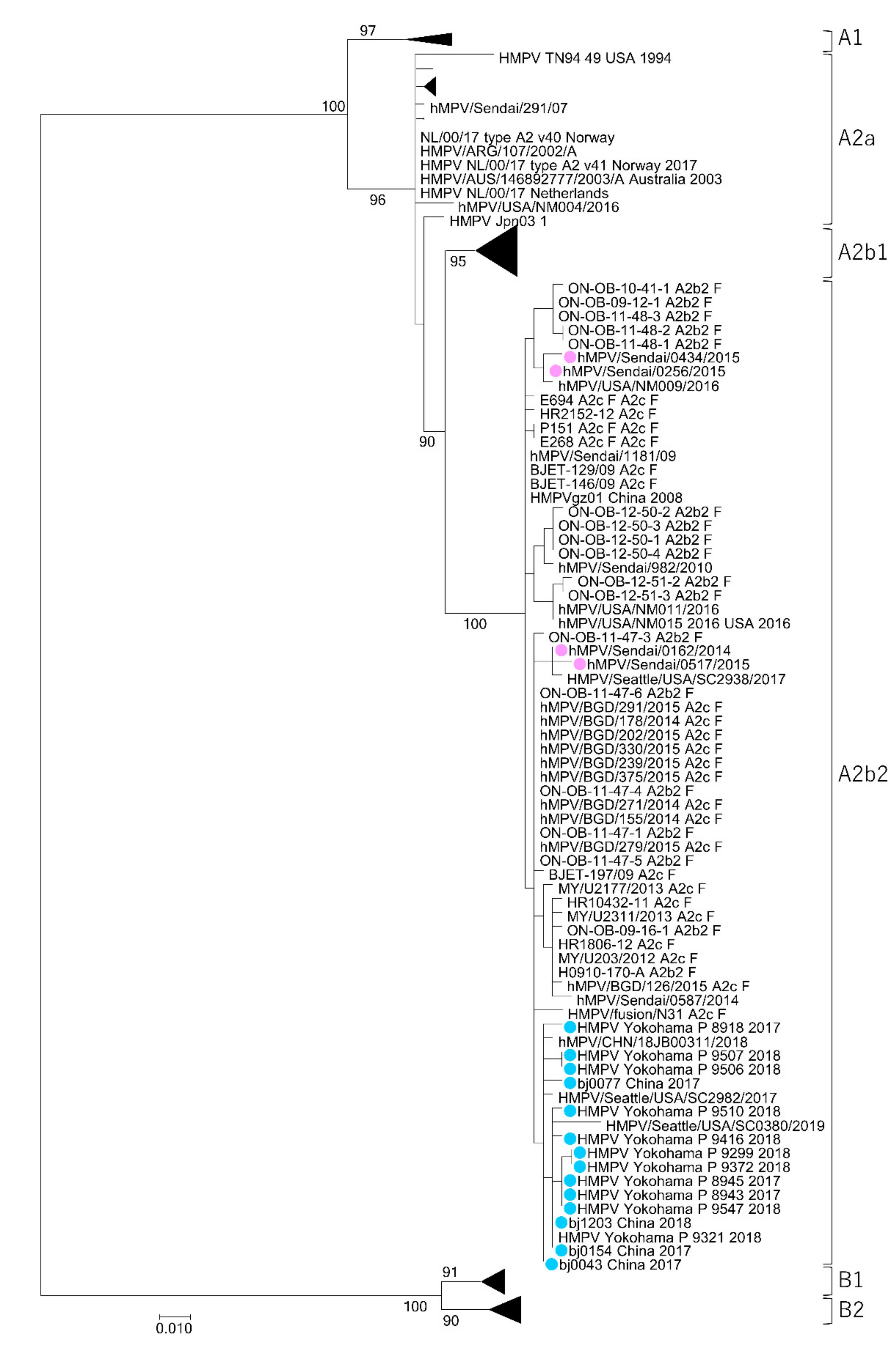
3.1. Isolation of HMPV A2b111nt-dup Strains and the Phylogenetic Relationship among HMPV Strains
3.2. Genetic Distances between HMPV Subtypes
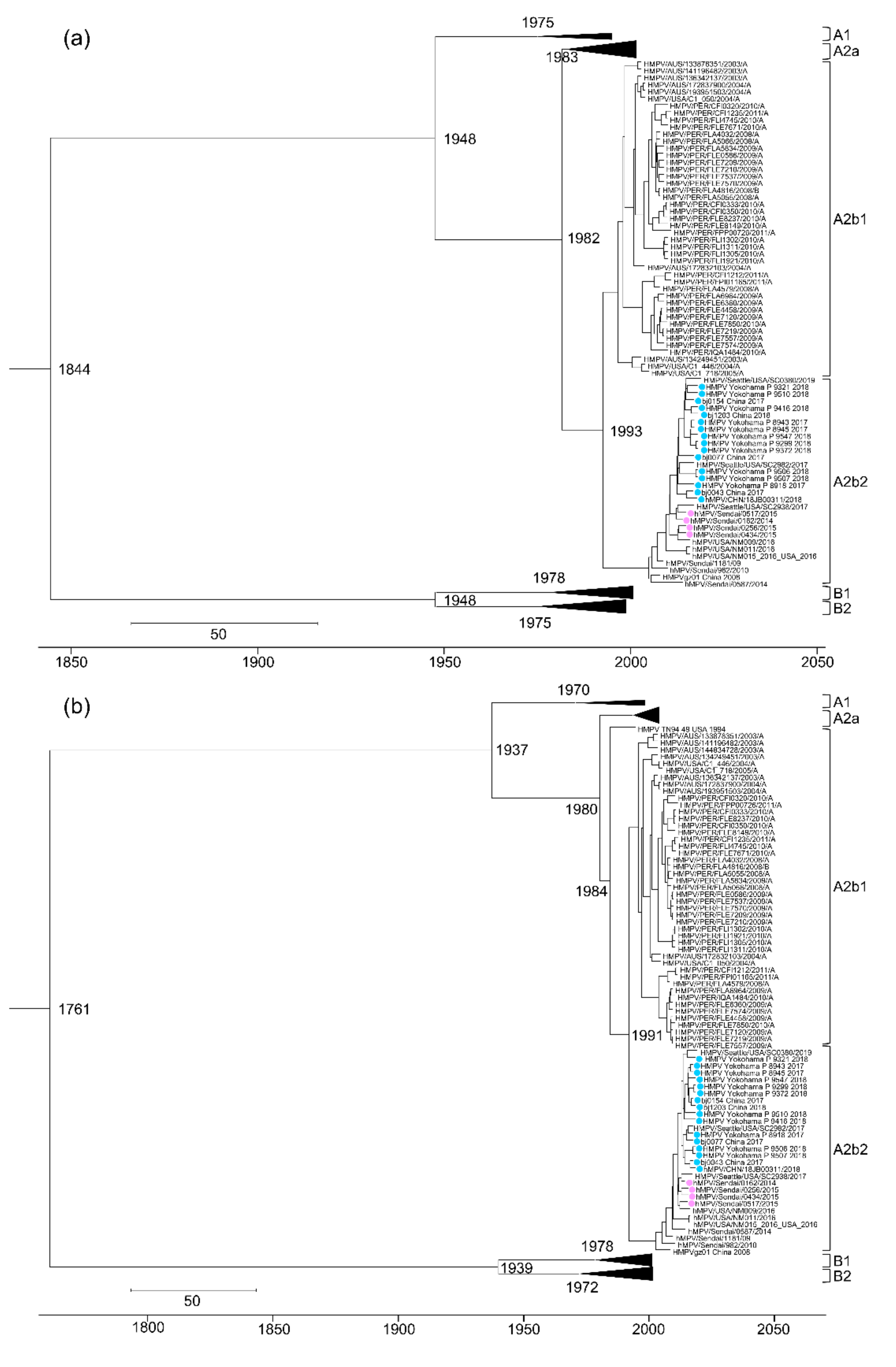
3.3. Evolutional Analysis of HMPV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Endo, R.; Kikuta, H.; Ishiguro, N.; Ishiko, H.; Hara, M.; Takahashi, Y.; Kobayashi, K. Human metapneumovirus infection in Japanese children. J. Clin. Microbiol. 2004, 42, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Abed, Y.; Pelletier, G.; Ruel, L.; Moisan, D.; Cote, S.; Peret, T.C.; Erdman, D.D.; Anderson, L.J. Virological features and clinical manifestations associated with human metapneumovirus: A new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J. Infect. Dis. 2002, 186, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Stockton, J.; Stephenson, I.; Fleming, D.; Zambon, M. Human metapneumovirus as a cause of community-acquired respiratory illness. Emerg. Infect. Dis. 2002, 8, 897–901. [Google Scholar] [CrossRef]
- Falsey, A.R.; Erdman, D.; Anderson, L.J.; Walsh, E.E. Human metapneumovirus infections in young and elderly adults. J. Infect. Dis. 2003, 187, 785–790. [Google Scholar] [CrossRef]
- Liao, R.S.; Appelgate, D.M.; Pelz, R.K. An outbreak of severe respiratory tract infection due to human metapneumovirus in a long-term care facility for the elderly in Oregon. J. Clin. Virol. 2012, 53, 171–173. [Google Scholar] [CrossRef]
- Louie, J.K.; Schnurr, D.P.; Pan, C.Y.; Kiang, D.; Carter, C.; Tougaw, S.; Ventura, J.; Norman, A.; Belmusto, V.; Rosenberg, J.; et al. A summer outbreak of human metapneumovirus infection in a long-term-care facility. J. Infect. Dis. 2007, 196, 705–708. [Google Scholar] [CrossRef]
- Biacchesi, S.; Skiadopoulos, M.H.; Yang, L.; Lamirande, E.W.; Tran, K.C.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Recombinant human Metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: Deletion of G yields a promising vaccine candidate. J. Virol. 2004, 78, 12877–12887. [Google Scholar] [CrossRef]
- Biacchesi, S.; Pham, Q.N.; Skiadopoulos, M.H.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 2005, 79, 12608–12613. [Google Scholar] [CrossRef]
- Chang, A.; Masante, C.; Buchholz, U.J.; Dutch, R.E. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J. Virol. 2012, 86, 3230–3243. [Google Scholar] [CrossRef]
- Bao, X.; Kolli, D.; Liu, T.; Shan, Y.; Garofalo, R.P.; Casola, A. Human metapneumovirus small hydrophobic protein inhibits NF-kappaB transcriptional activity. J. Virol. 2008, 82, 8224–8229. [Google Scholar] [CrossRef] [PubMed]
- Masante, C.; El Najjar, F.; Chang, A.; Jones, A.; Moncman, C.L.; Dutch, R.E. The human metapneumovirus small hydrophobic protein has properties consistent with those of a viroporin and can modulate viral fusogenic activity. J. Virol. 2014, 88, 6423–6433. [Google Scholar] [CrossRef] [PubMed]
- Adamson, P.; Thammawat, S.; Muchondo, G.; Sadlon, T.; Gordon, D. Diversity in glycosaminoglycan binding amongst hMPV G protein lineages. Viruses 2012, 4, 3785–3803. [Google Scholar] [CrossRef] [PubMed]
- Thammawat, S.; Sadlon, T.A.; Hallsworth, P.G.; Gordon, D.L. Role of cellular glycosaminoglycans and charged regions of viral G protein in human metapneumovirus infection. J. Virol. 2008, 82, 11767–11774. [Google Scholar] [CrossRef]
- Bao, X.; Liu, T.; Shan, Y.; Li, K.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS Pathog. 2008, 4, e1000077. [Google Scholar] [CrossRef]
- Van den Hoogen, B.G.; Herfst, S.; Sprong, L.; Cane, P.A.; Forleo-Neto, E.; de Swart, R.L.; Osterhaus, A.D.; Fouchier, R.A. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 2004, 10, 658–666. [Google Scholar] [CrossRef]
- Peret, T.C.T.; Abed, Y.; Anderson, L.J.; Erdman, D.D.; Boivin, G. Sequence polymorphism of the predicted human metapneumovirus G glycoprotein. J. Gen. Virol. 2004, 85, 679–686. [Google Scholar] [CrossRef]
- Biacchesi, S.; Skiadopoulos, M.H.; Boivin, G.; Hanson, C.T.; Murphy, B.R.; Collins, P.L.; Buchholz, U.J. Genetic diversity between human metapneumovirus subgroups. Virology 2003, 315, 1–9. [Google Scholar] [CrossRef]
- Huck, B.; Scharf, G.; Neumann-Haefelin, D.; Puppe, W.; Weigl, J.; Falcone, V. Novel human metapneumovirus sublineage. Emerg. Infect. Dis. 2006, 12, 147–150. [Google Scholar] [CrossRef]
- Saikusa, M.; Nao, N.; Kawakami, C.; Usuku, S.; Sasao, T.; Toyozawa, T.; Takeda, M.; Okubo, I. A novel 111-nucleotide duplication in the G gene of human metapneumovirus. Microbiol. Immunol. 2017, 61, 507–512. [Google Scholar] [CrossRef]
- Saikusa, M.; Kawakami, C.; Nao, N.; Takeda, M.; Usuku, S.; Sasao, T.; Nishimoto, K.; Toyozawa, T. 180-Nucleotide Duplication in the G Gene of Human metapneumovirus A2b Subgroup Strains Circulating in Yokohama City, Japan, since 2014. Front. Microbiol. 2017, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Nidaira, M.; Taira, K.; Hamabata, H.; Kawaki, T.; Gushi, K.; Mahoe, Y.; Maeshiro, N.; Azama, Y.; Okano, S.; Kyan, H.; et al. Molecular epidemiology of human metapneumovirus from 2009 to 2011 in Okinawa, Japan. Jpn. J. Infect. Dis. 2012, 65, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Regev, L.; Meningher, T.; Hindiyeh, M.; Mendelson, E.; Mandelboim, M. Increase human metapneumovirus mediated morbidity following pandemic influenza infection. PLoS ONE 2012, 7, e34750. [Google Scholar] [CrossRef] [PubMed]
- Nao, N.; Sato, K.; Yamagishi, J.; Tahara, M.; Nakatsu, Y.; Seki, F.; Katoh, H.; Ohnuma, A.; Shirogane, Y.; Hayashi, M.; et al. Consensus and variations in cell line specificity among human metapneumovirus strains. PLoS ONE 2019, 14, e0215822. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Gall, A.; Ong, S.H.; Brener, J.; Ferns, B.; Goulder, P.; Nastouli, E.; Keane, J.A.; Kellam, P.; Otto, T.D. IVA: Accurate de novo assembly of RNA virus genomes. Bioinformatics 2015, 31, 2374–2376. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Pinana, M.; Vila, J.; Gimferrer, L.; Valls, M.; Andres, C.; Codina, M.G.; Ramon, J.; Martin, M.C.; Fuentes, F.; Saiz, R.; et al. Novel human metapneumovirus with a 180-nucleotide duplication in the G gene. Future Microbiol. 2017, 12, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Saikusa, M.; Nao, N.; Kawakami, C.; Usuku, S.; Tanaka, N.; Tahara, M.; Takeda, M.; Okubo, I. Predominant Detection of the Subgroup A2b Human Metapneumovirus Strain with a 111-Nucleotide Duplication in the G gene in Yokohama City, Japan in 2018. Jpn. J. Infect. Dis. 2019, 72, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.Z.; Sumiya, M.; Sahabuddin, M.; Pell, L.G.; Gubbay, J.B.; Rahman, R.; Momtaz, F.; Azmuda, N.; Shanta, S.S.; Jahan, I.; et al. Genetic characterization of human metapneumovirus identified through community and facility-based surveillance of infants in Dhaka, Bangladesh. J. Med. Virol. 2019, 91, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Jallow, M.M.; Fall, A.; Kiori, D.; Sy, S.; Goudiaby, D.; Barry, M.A.; Fall, M.; Niang, M.N.; Dia, N. Epidemiological, clinical and genotypic features of human Metapneumovirus in patients with influenza-like illness in Senegal, 2012 to 2016. BMC Infect. Dis. 2019, 19, 457. [Google Scholar] [CrossRef]
- Jagusic, M.; Slovic, A.; Ljubin-Sternak, S.; Mlinaric-Galinovic, G.; Forcic, D. Genetic diversity of human metapneumovirus in hospitalized children with acute respiratory infections in Croatia. J. Med. Virol. 2017, 89, 1885–1893. [Google Scholar] [CrossRef]
- Neemuchwala, A.; Duvvuri, V.R.; Marchand-Austin, A.; Li, A.; Gubbay, J.B. Human metapneumovirus prevalence and molecular epidemiology in respiratory outbreaks in Ontario, Canada. J. Med. Virol. 2015, 87, 269–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Kim, J.I.; Park, S.; Lee, I.; Park, K.S.; Kwak, E.J.; Moon, K.M.; Lee, C.K.; Bae, J.Y.; Park, M.S.; Song, K.J. Genome-Wide Analysis of Human Metapneumovirus Evolution. PLoS ONE 2016, 11, e0152962. [Google Scholar] [CrossRef]
- Zhu, R.; Guo, C.; Zhao, L.; Deng, J.; Wang, F.; Sun, Y.; Qian, Y. Epidemiological and genetic characteristics of human metapneumovirus in pediatric patients across six consecutive seasons in Beijing, China. Int. J. Infect. Dis. 2020, 91, 137–142. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Isolation Year | Isolation Cell | 111nt-dup |
|---|---|---|---|
| HMPV/Yokohama/P8918/2017 | 2017 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P8943/2017 | 2017 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P8945/2017 | 2017 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9299/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9321/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9372/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9416/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9506/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9507/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9510/2018 | 2018 | VeroE6/TMPRSS2 | + |
| HMPV/Yokohama/P9547/2018 | 2018 | VeroE6/TMPRSS2 | + |
| - | Genetic Distance (p-Value) * | - | - | - | ||
|---|---|---|---|---|---|---|
| Subtype | A1 | A2a | A2b1 | A2b2 | B1 | B2 |
| A1 | 0.017 | |||||
| A2a | 0.076 | 0.013 | ||||
| A2b1 | 0.077 | 0.043 | 0.016 | |||
| A2b2 | 0.079 | 0.047 | 0.036 | 0.011 | ||
| B1 | 0.225 | 0.225 | 0.226 | 0.224 | 0.018 | |
| B2 | 0.226 | 0.225 | 0.227 | 0.225 | 0.073 | 0.017 |
| Subtypes | tMRCAs of Each Viral Gene | |||||||
|---|---|---|---|---|---|---|---|---|
| N | P | M | F | M2 | SH | G | L | |
| All | 1827 | 1775 | 1812 | 1844 | 1859 | 1645 | 1761 | 1754 |
| A | 1938 | 1937 | 1943 | 1948 | 1950 | 1937 | 1937 | 1925 |
| A1 | 1978 | 1972 | 1979 | 1975 | 1979 | 1972 | 1970 | 1973 |
| A2 | 1977 | 1978 | 1980 | 1982 | 1985 | 1971 | 1980 | 1977 |
| A2a | 1994 | 1993 | 1993 | 1994 | 1994 | 1989 | 1993 | 1992 |
| A2b | 1992 | 1991 | 1992 | 1993 | 1996 | 1980 | 1991 | 1989 |
| A2b1 | 1994 | 1994 | 1996 | 1997 | 1998 | 1991 | 1996 | 1995 |
| A2b2 | 2003 | 2003 | 2004 | 2005 | 2005 | 2004 | 2002 | 2002 |
| B | 1945 | 1924 | 1940 | 1948 | 1948 | 1934 | 1939 | 1933 |
| B1 | 1979 | 1984 | 1979 | 1975 | 1982 | 1969 | 1978 | 1980 |
| B2 | 1978 | 1950 | 1972 | 1978 | 1975 | 1969 | 1972 | 1969 |
| - | The Current Study | Regev et al. | Nidaira et al. |
|---|---|---|---|
| Reference | - | [23] | [22] |
| The number of viruses analyzed | 161 strains | 26 strains | 41 strains |
| Analyzed region | Full genome sequences (13 kb) | Partial F gene sequences (111 b) | Partial F gene sequences (321 b) |
| Phylogenetic analysis | Maximum likelihood analysis (IQ-TREE 2) | Nearest neighbor joint analysis (Clustal X) | Neighbor-joining analysis (MEGA) |
| Molecular clock phylogenies | Bayesian MCMC approach (BEAST2) | ND * | ND |
| Classification of HMPV A2 strains | A2a, A2b1, and A2b2 | A2a, A2b1, and A2b2 | A2a, A2b, and A2c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nao, N.; Saikusa, M.; Sato, K.; Sekizuka, T.; Usuku, S.; Tanaka, N.; Nishimura, H.; Takeda, M. Recent Molecular Evolution of Human Metapneumovirus (HMPV): Subdivision of HMPV A2b Strains. Microorganisms 2020, 8, 1280. https://doi.org/10.3390/microorganisms8091280
Nao N, Saikusa M, Sato K, Sekizuka T, Usuku S, Tanaka N, Nishimura H, Takeda M. Recent Molecular Evolution of Human Metapneumovirus (HMPV): Subdivision of HMPV A2b Strains. Microorganisms. 2020; 8(9):1280. https://doi.org/10.3390/microorganisms8091280
Chicago/Turabian StyleNao, Naganori, Miwako Saikusa, Ko Sato, Tsuyoshi Sekizuka, Shuzo Usuku, Nobuko Tanaka, Hidekazu Nishimura, and Makoto Takeda. 2020. "Recent Molecular Evolution of Human Metapneumovirus (HMPV): Subdivision of HMPV A2b Strains" Microorganisms 8, no. 9: 1280. https://doi.org/10.3390/microorganisms8091280
APA StyleNao, N., Saikusa, M., Sato, K., Sekizuka, T., Usuku, S., Tanaka, N., Nishimura, H., & Takeda, M. (2020). Recent Molecular Evolution of Human Metapneumovirus (HMPV): Subdivision of HMPV A2b Strains. Microorganisms, 8(9), 1280. https://doi.org/10.3390/microorganisms8091280