The Absence of C-5 DNA Methylation in Leishmania donovani Allows DNA Enrichment from Complex Samples

 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. In Silico Identification and Phylogeny of Putative DNMTs
2.2. Culturing and DNA Extraction for Bisulfite Sequencing
2.3. Genetic Engineering of L. donovani BPK282
2.4. Bisulfite Sequencing and Data Analysis
2.5. Leishmania DNA Enrichment from A Mix of Human and Leishmania DNA
3. Results
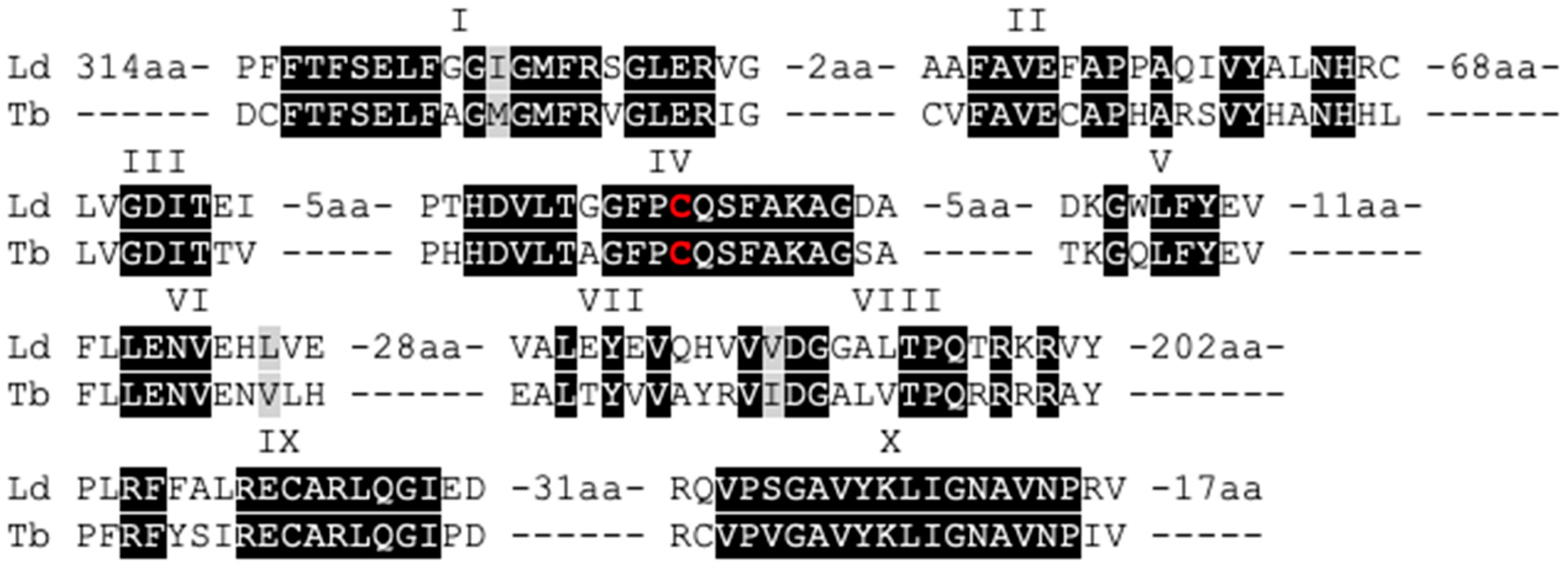
3.1. The Leishmania Genome Contains a Putative C-5 DNA Methyltransferase (DNMT)
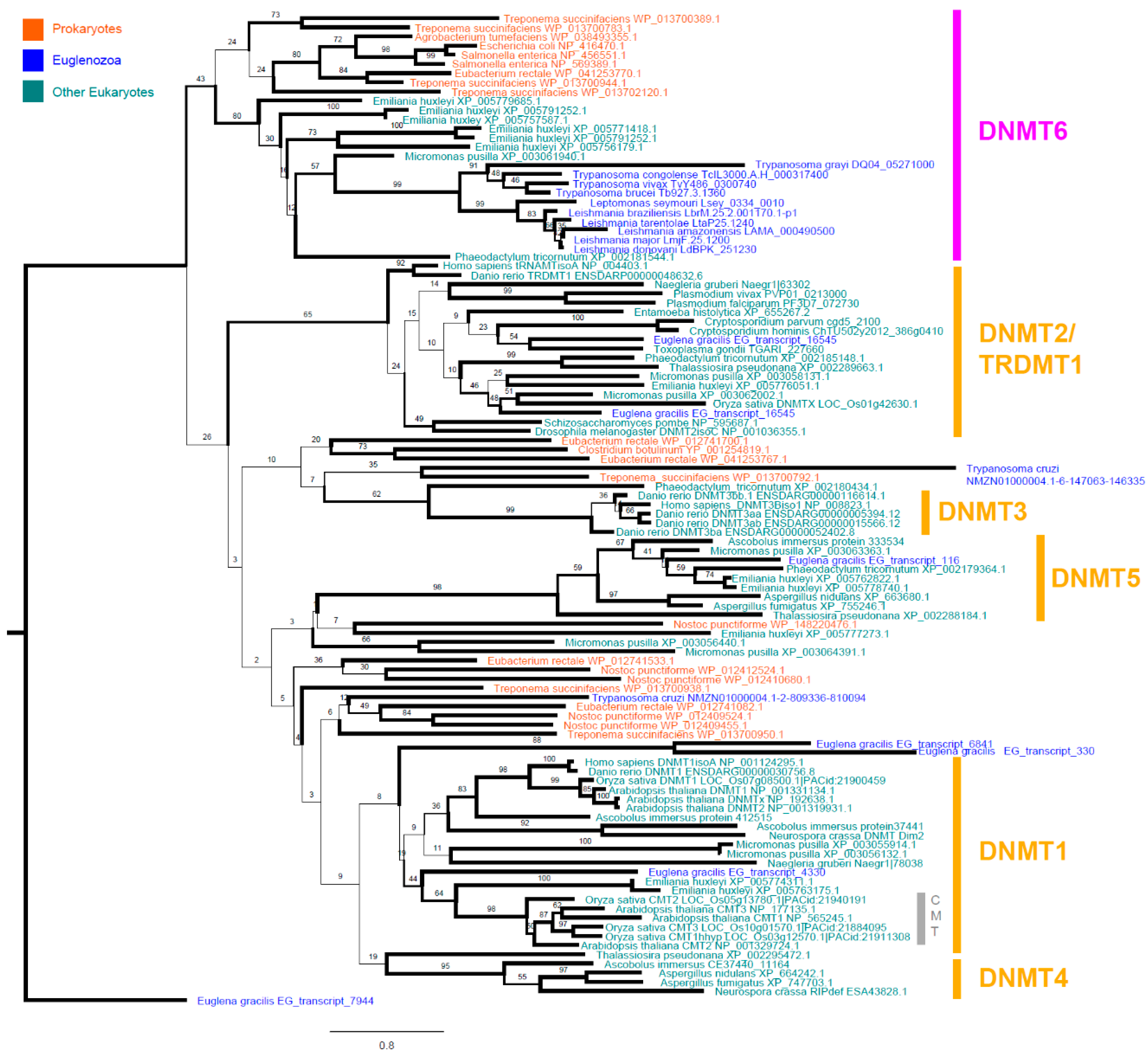
3.2. Leishmania and Trypanosomatid C-5 DNA Methyltransferase Belongs to the Eukaryotic DNMT6 Family
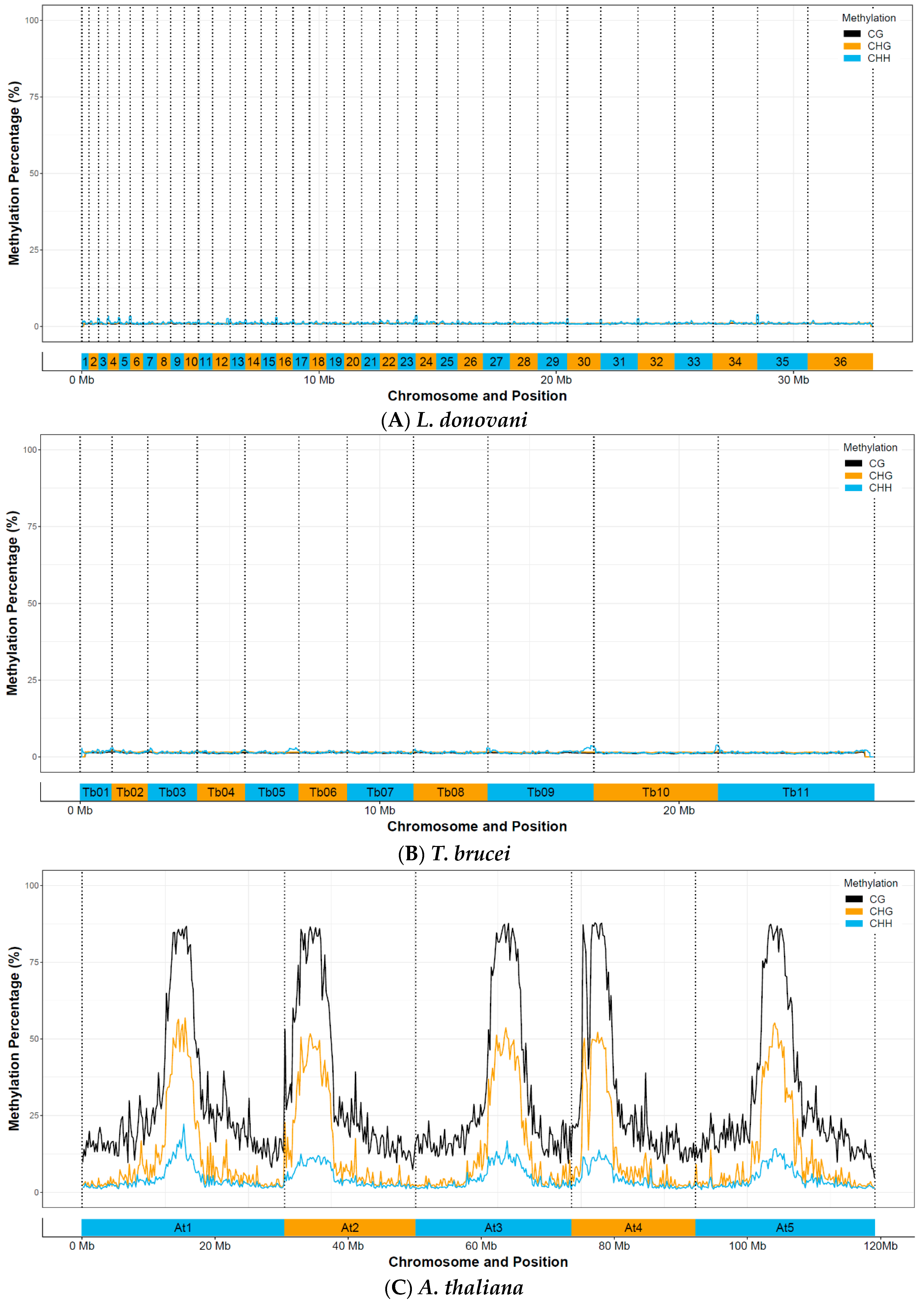
3.3. Whole-Genome Bisulfite Sequencing Reveals No Evidence for Functional C-5 Methylation
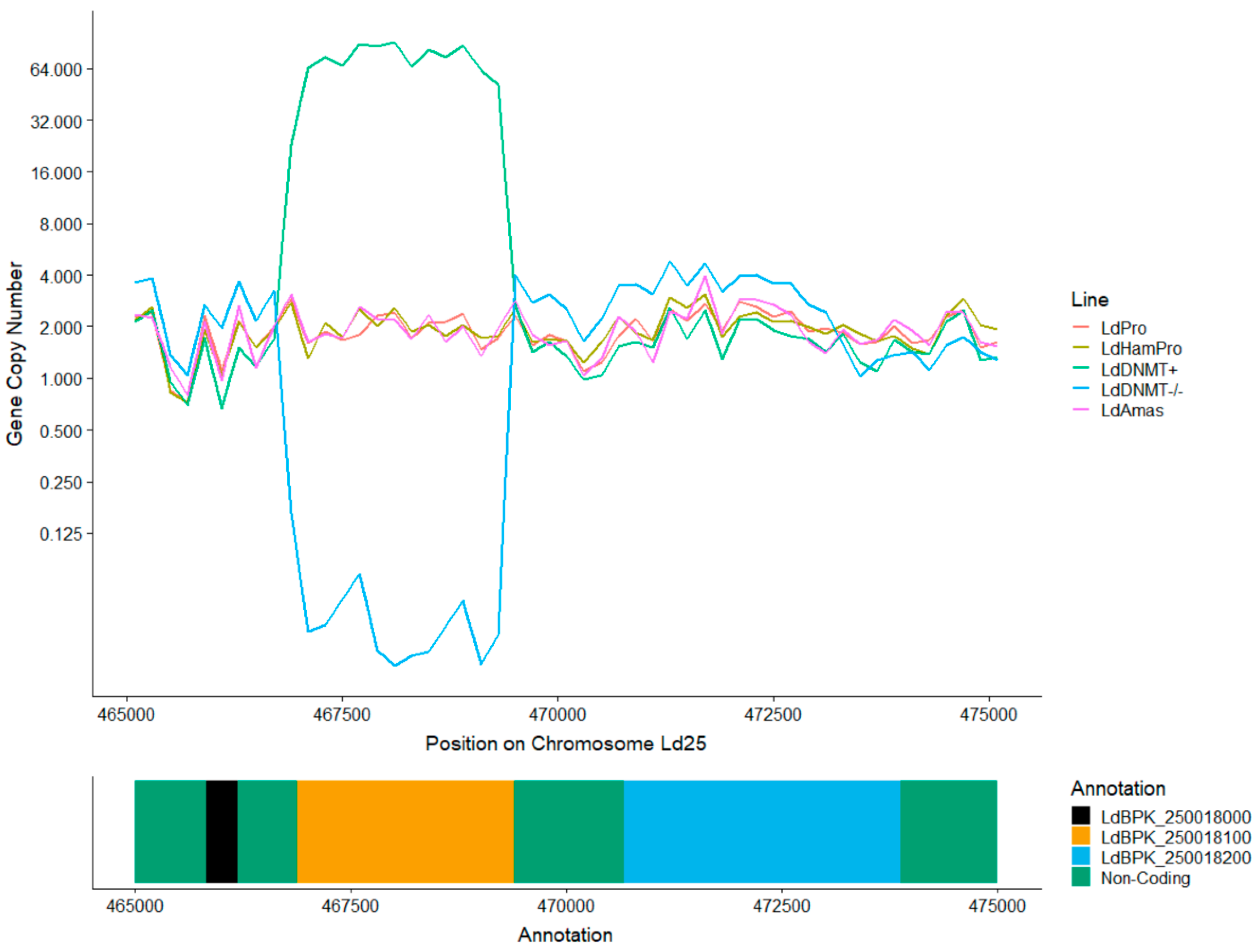
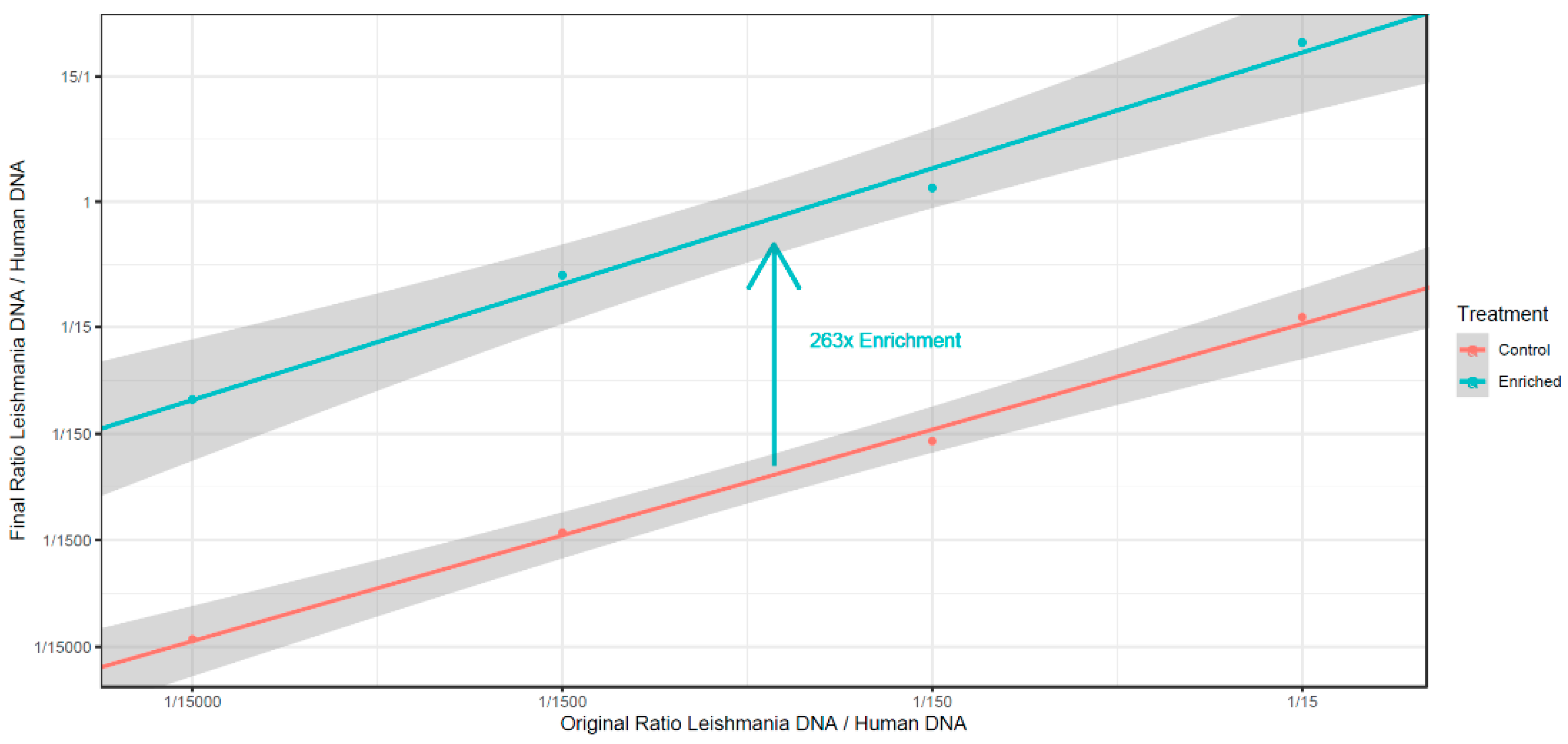
3.4. Absence of C5 DNA Methylation as a Leishmania vs. Host DNA Enrichment Strategy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jurkowski, T.P.; Jeltsch, A. On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PLoS ONE 2011, 6, e28104. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.; Sayols, S.; Moran, S.; Guillaumet-Adkins, A.; Schroeder, M.P.; Royo, R.; Orozco, M.; Gut, M.; Gut, I.; Lopez-Bigas, N.; et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene 2017, 36, 5648. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chung, W.-Y.; Qian, M.; Pellegrini, M.; Zhang, M.Q. Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells. Nucleic Acids Res. 2013, 42, 3009–3016. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Tomizawa, S.; Kobayashi, H.; Watanabe, T.; Andrews, S.; Hata, K.; Kelsey, G.; Sasaki, H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 2011, 138, 811–820. [Google Scholar] [CrossRef]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef]
- Chédin, F. Chapter 7-The DNMT3 family of mammalian de novo dna methyltransferases. In Progress in Molecular Biology and Translational Science; Cheng, X., Blumenthal, R.M., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 101, pp. 255–285. [Google Scholar]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef]
- Jurkowski, T.P.; Meusburger, M.; Phalke, S.; Helm, M.; Nellen, W.; Reuter, G.; Jeltsch, A. Human DNMT2 methylates tRNA(Asp) molecules using a DNA methyltransferase-like catalytic mechanism. RNA 2008, 14, 1663–1670. [Google Scholar] [CrossRef]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef]
- Burgess, A.L.; David, R.; Searle, I.R. Conservation of tRNA and rRNA 5-methylcytosine in the kingdom Plantae. BMC Plant Biol. 2015, 15, 199. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2016, 14, 1108–1123. [Google Scholar] [CrossRef]
- Ponts, N.; Fu, L.; Harris, E.Y.; Zhang, J.; Chung, D.-W.D.; Cervantes, M.C.; Prudhomme, J.; Atanasova-Penichon, V.; Zehraoui, E.; Bunnik, E.M.; et al. Genome-wide mapping of DNA methylation in the human malaria parasite Plasmodium falciparum. Cell Host Microbe 2013, 14, 696–706. [Google Scholar] [CrossRef]
- Motorin, Y.; Grosjean, H. Multisite-specific tRNA:m5C-methyltransferase (Trm4) in yeast Saccharomyces cerevisiae: Identification of the gene and substrate specificity of the enzyme. RNA 1999, 5, 1105–1118. [Google Scholar] [CrossRef]
- Jeltsch, A.; Nellen, W.; Lyko, F. Two substrates are better than one: Dual specificities for Dnmt2 methyltransferases. Trends Biochem. Sci. 2006, 31, 306–308. [Google Scholar] [CrossRef]
- Kaiser, S.; Jurkowski, T.P.; Kellner, S.; Schneider, D.; Jeltsch, A.; Helm, M. The RNA methyltransferase Dnmt2 methylates DNA in the structural context of a tRNA. RNA Biol. 2016, 14, 1241–1251. [Google Scholar] [CrossRef]
- Ponger, L.; Li, W.H. Evolutionary diversification of DNA methyltransferases in eukaryotic genomes. Mol. Biol. Evol. 2005, 22, 1119–1128. [Google Scholar] [CrossRef]
- Wei, H.; Jiang, S.; Chen, L.; He, C.; Wu, S.; Peng, H. Characterization of cytosine methylation and the DNA methyltransferases of toxoplasma gondii. Int. J. Biol. Sci. 2017, 13, 458–470. [Google Scholar] [CrossRef]
- Morselli, M.; Pastor, W.A.; Montanini, B.; Nee, K.; Ferrari, R.; Fu, K.; Bonora, G.; Rubbi, L.; Clark, A.T.; Ottonello, S.; et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. eLife 2015, 4, e06205. [Google Scholar] [CrossRef]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizábal-Corrales, D.; Hsu, C.-H.; Aravind, L.; He, C.; Shi, Y. DNA methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef]
- Van Luenen, H.G.A.M.; Farris, C.; Jan, S.; Genest, P.-A.; Tripathi, P.; Velds, A.; Kerkhoven, R.M.; Nieuwland, M.; Haydock, A.; Ramasamy, G.; et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell 2012, 150, 909–921. [Google Scholar] [CrossRef]
- Huff, J.T.; Zilberman, D. Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 2014, 156, 1286–1297. [Google Scholar] [CrossRef]
- Militello, K.T.; Wang, P.; Jayakar, S.K.; Pietrasik, R.L.; Dupont, C.D.; Dodd, K.; King, A.M.; Valenti, P.R. African trypanosomes contain 5-Methylcytosine in nuclear DNA. Eukaryot. Cell 2008, 7, 2012–2016. [Google Scholar] [CrossRef]
- Bateman, A.; Smart, A.; Luciani, A.; Salazar, G.A.; Mistry, J.; Richardson, L.J.; Qureshi, M.; El-Gebali, S.; Potter, S.C.; Finn, R.D.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef]
- Dumetz, F.; Imamura, H.; Sanders, M.; Seblova, V.; Myskova, J.; Pescher, P.; Vanaerschot, M.; Meehan, C.J.; Cuypers, B.; De Muylder, G.; et al. Modulation of aneuploidy in leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio 2017, 8, e00599-17. [Google Scholar] [CrossRef]
- Real, F.; Vidal, R.O.; Carazzolle, M.F.; Mondego, J.M.C.; Costa, G.G.L.; Herai, R.H.; Würtele, M.; de Carvalho, L.M.; Carmona e Ferreira, R.; Mortara, R.A.; et al. The genome sequence of Leishmania (Leishmania) amazonensis: Functional annotation and extended analysis of gene models. DNA Res. 2013, 20, 567–581. [Google Scholar] [CrossRef]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef]
- Raymond, F.; Boisvert, S.; Roy, G.; Ritt, J.-F.; Légaré, D.; Isnard, A.; Stanke, M.; Olivier, M.; Tremblay, M.J.; Papadopoulou, B.; et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012, 40, 1131–1147. [Google Scholar] [CrossRef]
- Kraeva, N.; Butenko, A.; Hlaváčová, J.; Kostygov, A.; Myškova, J.; Grybchuk, D.; Leštinová, T.; Votýpka, J.; Volf, P.; Opperdoes, F.; et al. Leptomonas seymouri: Adaptations to the dixenous life cycle analyzed by genome sequencing, transcriptome profiling and co-infection with leishmania donovani. PLoS Pathog. 2015, 11, e1005127. [Google Scholar] [CrossRef]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef]
- Abbas, A.H.; Silva Pereira, S.; D’Archivio, S.; Wickstead, B.; Morrison, L.J.; Hall, N.; Hertz-Fowler, C.; Darby, A.C.; Jackson, A.P. The structure of a conserved telomeric region associated with variant antigen loci in the blood parasite trypanosoma congolense. Genome Biol. Evol. 2018, 10, 2458–2473. [Google Scholar] [CrossRef]
- Kelly, S.; Ivens, A.; Manna, P.T.; Gibson, W.; Field, M.C. A draft genome for the African crocodilian trypanosome Trypanosoma grayi. Sci. Data 2014, 1, 140024. [Google Scholar] [CrossRef]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–D543. [Google Scholar] [CrossRef] [PubMed]
- Auburn, S.; Böhme, U.; Steinbiss, S.; Trimarsanto, H.; Hostetler, J.; Sanders, M.; Gao, Q.; Nosten, F.; Newbold, C.I.; Berriman, M.; et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 2016, 1, 4. [Google Scholar] [CrossRef]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Puiu, D.; Enomoto, S.; Buck, G.A.; Abrahamsen, M.S.; Kissinger, J.C. CryptoDB: The Cryptosporidium genome resource. Nucleic Acids Res. 2004, 32, D329–D331. [Google Scholar] [CrossRef]
- Abrahamsen, M.S.; Templeton, T.J.; Enomoto, S.; Abrahante, J.E.; Zhu, G.; Lancto, C.A.; Deng, M.; Liu, C.; Widmer, G.; Tzipori, S.; et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 2004, 304, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Widmer, G.; Wang, Y.; Ozaki, L.S.; Alves, J.M.; Serrano, M.G.; Puiu, D.; Manque, P.; Akiyoshi, D.; Mackey, A.J.; et al. The genome of Cryptosporidium hominis. Nature 2004, 431, 1107–1112. [Google Scholar] [CrossRef]
- Gajria, B.; Bahl, A.; Brestelli, J.; Dommer, J.; Fischer, S.; Gao, X.; Heiges, M.; Iodice, J.; Kissinger, J.C.; Mackey, A.J.; et al. ToxoDB: An integrated Toxoplasma gondii database resource. Nucleic Acids Res. 2008, 36, D553–D556. [Google Scholar] [CrossRef]
- Lorenzi, H.; Khan, A.; Behnke, M.S.; Namasivayam, S.; Swapna, L.S.; Hadjithomas, M.; Karamycheva, S.; Pinney, D.; Brunk, B.P.; Ajioka, J.W.; et al. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat. Commun. 2016, 7, 10147. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Cho, S.T.; Lo, W.S.; Wang, Y.C.; Lai, E.M.; Kuo, C.H. Complete genome sequence of agrobacterium tumefaciens Ach5. Genome Announc. 2015, 3, e00570-15. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.-J.; Wortman, J.R.; Batzoglou, S.; Lee, S.-I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Sebaihia, M.; Peck, M.W.; Minton, N.P.; Thomson, N.R.; Holden, M.T.; Mitchell, W.J.; Carter, A.T.; Bentley, S.D.; Mason, D.R.; Crossman, L.; et al. Genome sequence of a proteolytic (Group I) Clostridium botulinum strain Hall A and comparative analysis of the clostridial genomes. Genome Res. 2007, 17, 1082–1092. [Google Scholar] [CrossRef]
- Puerta, M.V.S.; Bachvaroff, T.R.; Delwiche, C.F. The complete plastid genome sequence of the haptophyte emiliania huxleyi: A comparison to other plastid genomes. DNA Res. 2005, 12, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, H.A.; Puiu, D.; Miller, J.R.; Brinkac, L.M.; Amedeo, P.; Hall, N.; Caler, E.V. New assembly, reannotation and analysis of the Entamoeba histolytica genome reveal new genomic features and protein content information. PLoS Negl. Trop. Dis. 2010, 4, e716. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, T.E.; Zoltner, M.; Burrell, A.; Nenarokova, A.; Novák Vanclová, A.M.G.; Prasad, B.; Soukal, P.; Santana-Molina, C.; O’Neill, E.; Nankissoor, N.N.; et al. Transcriptome, proteome and draft genome of Euglena gracilis. BMC Biol. 2019, 17, 11. [Google Scholar] [CrossRef]
- Worden, A.Z.; Lee, J.-H.; Mock, T.; Rouzé, P.; Simmons, M.P.; Aerts, A.L.; Allen, A.E.; Cuvelier, M.L.; Derelle, E.; Everett, M.V.; et al. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes micromonas. Science 2009, 324, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Parkhill, J.; Dougan, G.; James, K.D.; Thomson, N.R.; Pickard, D.; Wain, J.; Churcher, C.; Mungall, K.L.; Bentley, S.D.; Holden, M.T.; et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 2001, 413, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, J.; Alborn, W.E., Jr.; Arnold, J.; Blaszczak, L.C.; Burgett, S.; DeHoff, B.S.; Estrem, S.T.; Fritz, L.; Fu, D.J.; Fuller, W.; et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 2001, 183, 5709–5717. [Google Scholar] [CrossRef]
- Han, C.; Gronow, S.; Teshima, H.; Lapidus, A.; Nolan, M.; Lucas, S.; Hammon, N.; Deshpande, S.; Cheng, J.-F.; Zeytun, A.; et al. Complete genome sequence of Treponema succinifaciens type strain (6091). Stand. Genomic Sci. 2011, 4, 361–370. [Google Scholar] [CrossRef]
- Murat, C.; Payen, T.; Noel, B.; Kuo, A.; Morin, E.; Chen, J.; Kohler, A.; Krizsan, K.; Balestrini, R.; Da Silva, C.; et al. Pezizomycetes genomes reveal the molecular basis of ectomycorrhizal truffle lifestyle. Nat. Ecol. Evol. 2018, 2, 1956–1965. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Glez-Peña, D.; Gomez-Blanco, D.; Reboiro-Jato, M.; Fdez-Riverola, F.; Posada, D. ALTER: Program-oriented conversion of DNA and protein alignments. Nucleic Acids Res. 2010, 38, W14–W18. [Google Scholar] [CrossRef] [PubMed]
- Pescher, P.; Blisnick, T.; Bastin, P.; Spath, G.F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cell. Microbiol. 2011, 13, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Tihon, E.; Imamura, H.; Dujardin, J.C.; Van Den Abbeele, J.; Van den Broeck, F. Discovery and genomic analyses of hybridization between divergent lineages of Trypanosoma congolense, causative agent of Animal African Trypanosomiasis. Mol. Ecol. 2017, 26, 6524–6538. [Google Scholar] [CrossRef] [PubMed]
- LeBowitz, J.H. Transfection experiments with Leishmania. Methods Cell Biol. 1994, 45, 65–78. [Google Scholar] [PubMed]
- Morales, M.A.; Watanabe, R.; Dacher, M.; Chafey, P.; Osorio y Fortea, J.; Scott, D.A.; Beverley, S.M.; Ommen, G.; Clos, J.; Hem, S.; et al. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc. Natl. Acad. Sci. USA 2010, 107, 8381–8386. [Google Scholar] [CrossRef]
- Dumetz, F.; Cuypers, B.; Imamura, H.; Zander, D.; D’Haenens, E.; Maes, I.; Domagalska, M.A.; Clos, J.; Dujardin, J.C.; De Muylder, G. Molecular preadaptation to antimony resistance in leishmania donovani on the Indian subcontinent. mSphere 2018, 3, e00548-17. [Google Scholar] [CrossRef]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.-Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef]
- Liao, W.W.; Yen, M.R.; Ju, E.; Hsu, F.M.; Lam, L.; Chen, P.Y. MethGo: A comprehensive tool for analyzing whole-genome bisulfite sequencing data. BMC Genomics 2015, 16 (Suppl. S12), 1–8. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Decuypere, S.; Vanaerschot, M.; Rijal, S.; Yardley, V.; Maes, L.; de Doncker, S.; Chappuis, F.; Dujardin, J.C. Gene expression profiling of Leishmania (Leishmania) donovani: Overcoming technical variation and exploiting biological variation. Parasitology 2008, 135, 183–194. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Imamura, H.; Sanders, M.; Van den Broeck, F.; Bhattarai, N.R.; Vanaerschot, M.; Maes, I.; D’Haenens, E.; Rai, K.; Rijal, S.; et al. Genomes of intracellular Leishmania parasites directly sequenced from patients. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cao, X.; Aufsatz, W.; Zilberman, D.; Mette, M.F.; Huang, M.S.; Matzke, M.; Jacobsen, S.E. Role of the DRM and CMT3 methyltransferases in RNA-Directed DNA methylation. Curr. Biol. 2003, 13, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- De Mendoza, A.; Bonnet, A.; Vargas-Landin, D.B.; Ji, N.; Li, H.; Yang, F.; Li, L.; Hori, K.; Pflueger, J.; Buckberry, S.; et al. Recurrent acquisition of cytosine methyltransferases into eukaryotic retrotransposons. Nat. Commun. 2018, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.S.; Osborne, E.J.; Paolo Casale, F.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjalmsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, R.; Katara, G.K.; Singh, L.C.; Negi, N.S.; Ramesh, V.; Salotra, P. Quantification of parasite load in clinical samples of leishmaniasis patients: IL-10 level correlates with parasite load in visceral leishmaniasis. PLoS ONE 2010, 5, e10107. [Google Scholar] [CrossRef]
- Cuypers, B.; Berg, M.; Imamura, H.; Dumetz, F.; De Muylder, G.; Domagalska, M.A.; Rijal, S.; Bhattarai, N.R.; Maes, I.; Sanders, M. Integrated genomic and metabolomic profiling of ISC1, an emerging Leishmania donovani population in the Indian subcontinent. Infect. Genet. Evol. 2018, 62, 170–178. [Google Scholar] [CrossRef]
- Imamura, H.; Downing, T.; Van den Broeck, F.; Sanders, M.J.; Rijal, S.; Sundar, S.; Mannaert, A.; Vanaerschot, M.; Berg, M.; De Muylder, G.; et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. eLife 2016, 5, e12613. [Google Scholar] [CrossRef] [PubMed]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Vogel, K.J.; Moore, A.J.; Schmitz, R.J. The evolution of DNA methylation and its relationship to sociality in insects. bioRxiv 2016, 062455. [Google Scholar] [CrossRef]
- Oyola, S.O.; Gu, Y.; Manske, M.; Otto, T.D.; O’Brien, J.; Alcock, D.; Macinnis, B.; Berriman, M.; Newbold, C.I.; Kwiatkowski, D.P.; et al. Efficient depletion of host DNA contamination in malaria clinical sequencing. J. Clin. Microbiol. 2013, 51, 745–751. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CpG (%) | CHG (%) | CHH (%) | |
|---|---|---|---|
| LdPro | 0.0003 | 0.0005 | 0.0126 |
| LdAmas | 0.0001 | 0.0003 | 0.0073 |
| LdHamPro | 0.0002 | 0.0005 | 0.0113 |
| LdDNMT+ | 0.0013 | 0.0026 | 0.0627 |
| LdDNMT-/- | 0.0002 | 0.0006 | 0.0079 |
| Tbrucei | 0.0001 | 0.0006 | 0.0040 |
| Athaliana | 21.0473 | 4.0401 | 0.3141 |
| Ldo-Pro | LdDNMT+ | |
|---|---|---|
| RNA | 1.53 ± 0.2 | 3.78 ± 0.3 |
| BPK026 | BPK275 | BPK282 | Average Enrichment (X) | St.Dev | |
|---|---|---|---|---|---|
| Promastigotes | 79.85 | 88.32 | 60.47 | 76.22 | 14.28 |
| Amastigotes | 64.83 | 56.87 | 63.33 | 61.68 | 4.23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuypers, B.; Dumetz, F.; Meysman, P.; Laukens, K.; De Muylder, G.; Dujardin, J.-C.; Domagalska, M.A. The Absence of C-5 DNA Methylation in Leishmania donovani Allows DNA Enrichment from Complex Samples. Microorganisms 2020, 8, 1252. https://doi.org/10.3390/microorganisms8081252
Cuypers B, Dumetz F, Meysman P, Laukens K, De Muylder G, Dujardin J-C, Domagalska MA. The Absence of C-5 DNA Methylation in Leishmania donovani Allows DNA Enrichment from Complex Samples. Microorganisms. 2020; 8(8):1252. https://doi.org/10.3390/microorganisms8081252
Chicago/Turabian StyleCuypers, Bart, Franck Dumetz, Pieter Meysman, Kris Laukens, Géraldine De Muylder, Jean-Claude Dujardin, and Malgorzata Anna Domagalska. 2020. "The Absence of C-5 DNA Methylation in Leishmania donovani Allows DNA Enrichment from Complex Samples" Microorganisms 8, no. 8: 1252. https://doi.org/10.3390/microorganisms8081252
APA StyleCuypers, B., Dumetz, F., Meysman, P., Laukens, K., De Muylder, G., Dujardin, J.-C., & Domagalska, M. A. (2020). The Absence of C-5 DNA Methylation in Leishmania donovani Allows DNA Enrichment from Complex Samples. Microorganisms, 8(8), 1252. https://doi.org/10.3390/microorganisms8081252