Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches

 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Bacteriophage Isolation and Purification
2.3. Host Range of Bacteriophages
2.4. Biological Activity of Bacteriophages
2.5. Electron Microscopy
2.6. Phage Genome Sequencing and Annotation
2.7. Phylogenetic Analysis
2.8. Whole-Genome and Proteome Analysis
2.9. D Homology Modelling, Alignment and Visualisation
3. Results
3.1. General Properties of Pectobacterium Bacteriophages PP47, PP81 and Q19
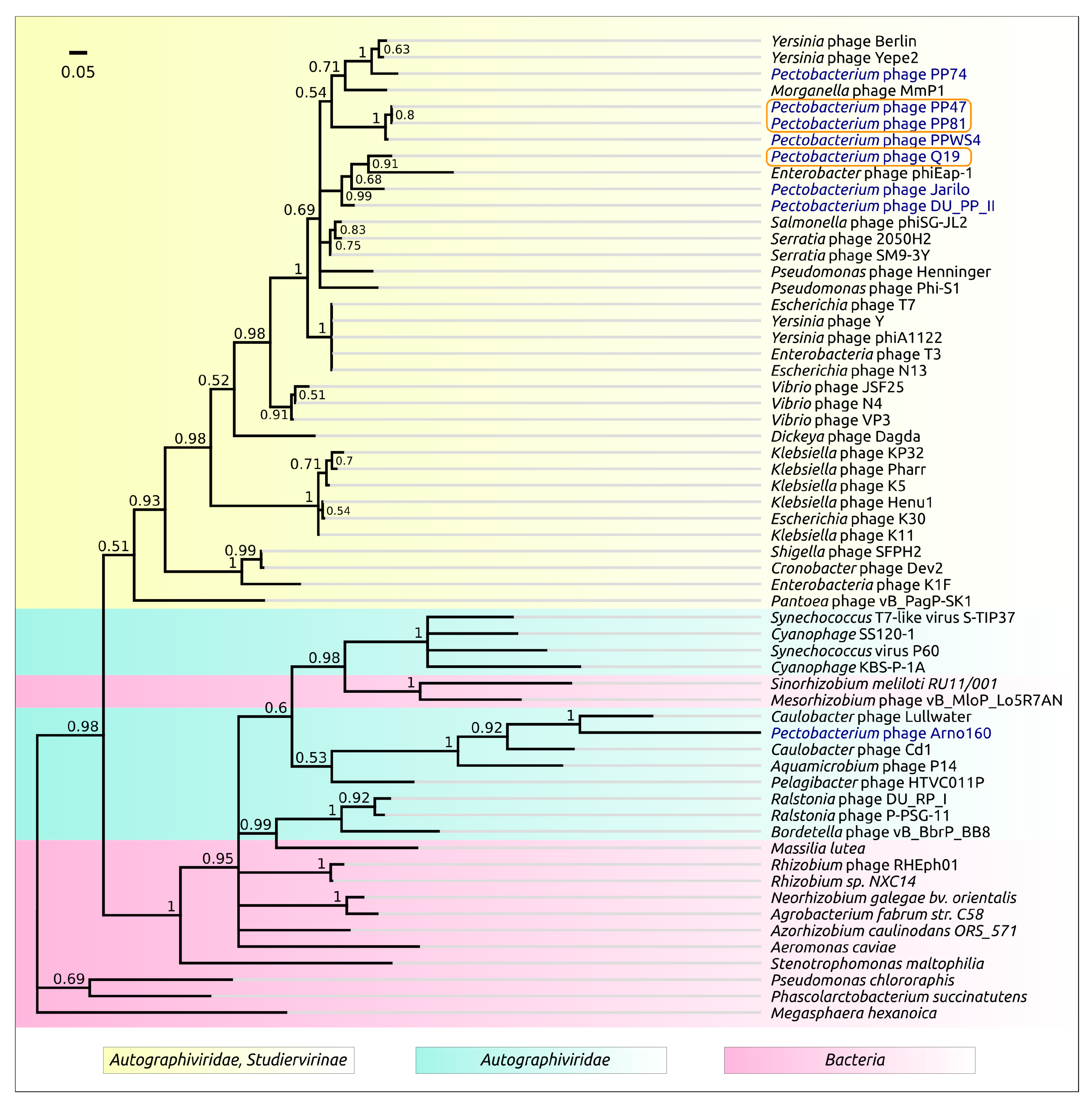
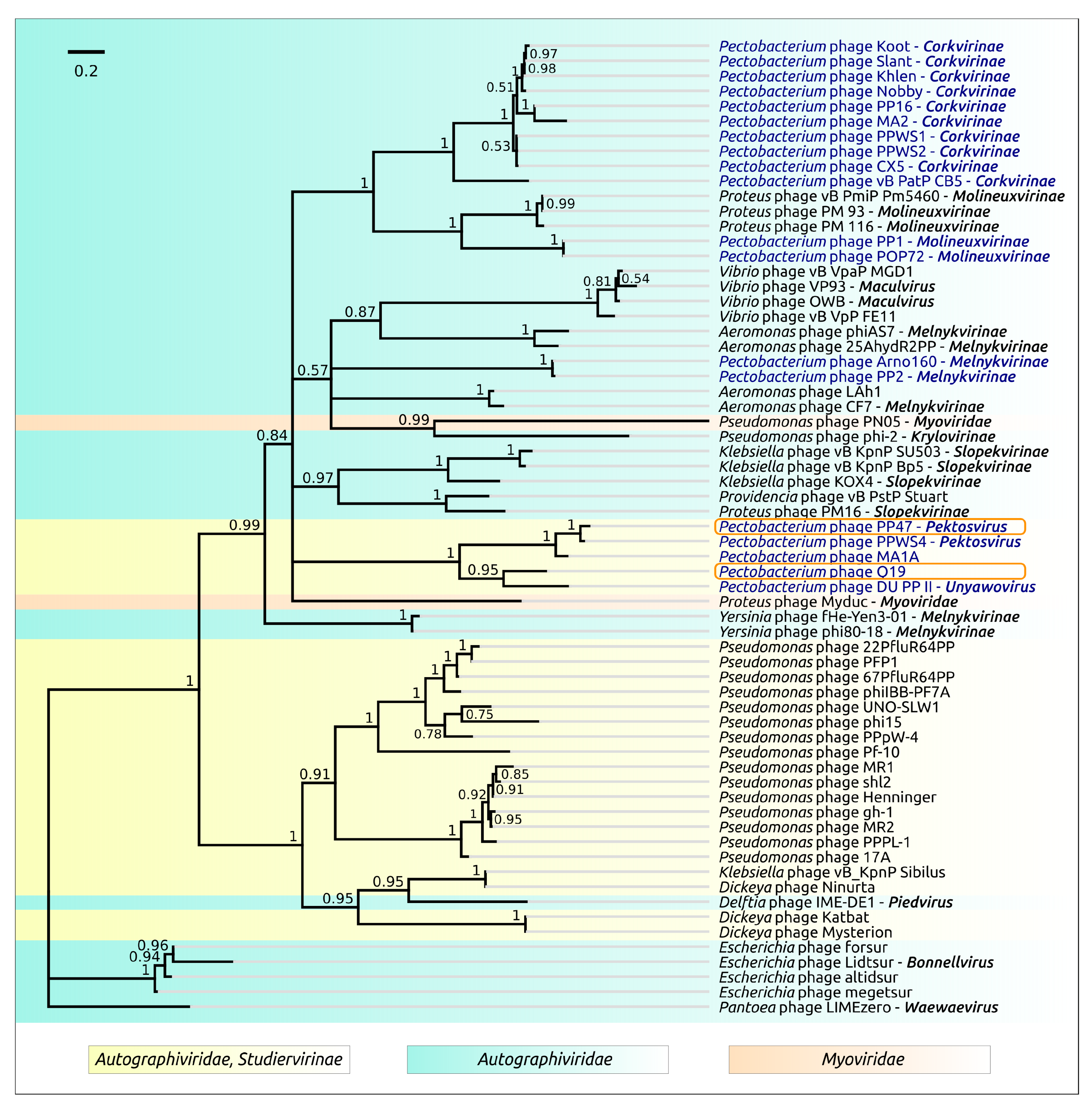
3.2. Taxonomy
3.3. Proteome Analysis
3.4. Genomic Analysis
3.5. Tail Spike Proteins
3.6. tRNA-Nucleotidyltransferase
4. Discussion
4.1. Origin, Phylogeny and Taxonomy
4.2. Genome, Adsorption Apparatus and Horizontal Transfer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Czajkowski, R.; Pérombelon, M.C.M.; Van Veen, J.A.; Van der Wolf, J.M. Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: A review. Plant. Pathol. 2011, 60, 999–1013. [Google Scholar] [CrossRef]
- Pérombelon, M.C.M. Potato diseases caused by soft rot erwinias: An overview of pathogenesis. Plant. Pathol. 2002, 51, 1–12. [Google Scholar]
- Ma, B.; Hibbing, M.E.; Kim, H.-S.; Reedy, R.M.; Yedidia, I.; Breuer, J.J.; Breuer, J.J.; Glasner, J.D.; Perna, N.T.; Kelman, A.; et al. Host range and molecular phylogenies of the soft rot enterobacterial genera pectobacterium and dickeya. Phytopathology 2007, 97, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Motyka, A.; Zoledowska, S.; Sledz, W.; Lojkowska, E. Molecular methods as tools to control plant diseases caused by Dickeya and Pectobacterium spp: A minireview. New Biotechnol. 2017, 39, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Svircev, A.; Roach, D.; Castle, A. Framing the future with bacteriophages in agriculture. Viruses 2018, 218. [Google Scholar] [CrossRef] [PubMed]
- Sillankorva, S.M.; Oliveira, H.; Azeredo, J. Bacteriophages and their role in food safety. Int. J. Microbiol. 2012, 2012, 863945. [Google Scholar] [CrossRef] [PubMed]
- Zaczek, M.; Weber-Dabrowska, B.; Górski, A. Phages in the global fruit and vegetable industry. J. Appl. Microbiol. 2015, 118, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; McAuliffe, O.; Ross, R.P.; Hill, C.; O’Mahony, J.; Coffey, A. Bacteriophages and Bacterial Plant Diseases. Front. Microbiol. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, R.; Ozymko, Z.; Lojkowska, E. Isolation and characterization of novel soilborne lytic bacteriophages infecting Dickeya spp. biovar 3 (‘D. solani’). Plant. Pathol. 2014, 63, 758–772. [Google Scholar] [CrossRef]
- Carstens, A.B.; Djurhuus, A.M.; Kot, W.; Hansen, L.H. A novel six-phage cocktail reduces Pectobacterium atrosepticum soft rot infection in potato tubers under simulated storage conditions. FEMS Microbiol. Lett. 2019, 366, i97–i104. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; van Vaerenbergh, J.; Vandenheuvel, D.; Dunon, V.; Ceyssens, P.J.; de Proft, M.; Kropinski, A.M.; Noben, J.P.; Maes, M.; Lavigne, R. T4-related bacteriophage LIMEstone isolates for the control of soft rot on potato caused by “Dickeya solani”. PLoS ONE 2012, 7, e33227. [Google Scholar] [CrossRef] [PubMed]
- Voronina, M.V.; Bugaeva, E.N.; Vasiliev, D.N.; Kabanova, A.P.; Barannik, A.P.; Shneider, M.M.; Kulikov, E.E.; Korzhenkov, A.A.; Toschakov, S.V.; Ignatov, A.N.; et al. Characterization of Pectobacterium carotovorum subsp. carotovorum Bacteriophage PP16 Prospective for Biocontrol of Potato Soft Rot. Mikrobiologiia 2019, 13, 458–469. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morgane. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, Q.; Loria, R. A re-evaluation of the taxonomy of phytopathogenic genera Dickeya and Pectobacterium using whole-genome sequencing data. Syst. Appl. Microbiol. 2016, 39, 252–259. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.J.; Kropinski, A.M. Bacteriophages: Methods and Protocols Volume 1: Isolation, Characterization, and Interactions; Humana Press: Leicester, UK, 2009; ISBN 9781588296825. [Google Scholar]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinform. 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Sampaio, M.; Rocha, M.; Oliveira, H.; DIas, O.; Valencia, A. Predicting promoters in phage genomes using PhagePromoter. Bioinformatics 2019, 35, 5301–5302. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Rappas, M.; Bose, D.; Zhang, X. Bacterial enhancer-binding proteins: Unlocking σ54-dependent gene transcription. Curr. Opin. Struct. Biol. 2007, 17, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Bioinformatics 1994, 10, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-a novel tool to calculate the intergenomic 1 similarities of prokaryote-infecting viruses 2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Duarte, V.; De Boer, S.H.; Ward, L.J.; De Oliveira, A.M.R. Characterization of atypical Erwinia carotovora strains causing blackleg of potato in Brazil. J. Appl. Microbiol. 2004, 96, 535–545. [Google Scholar] [CrossRef]
- Portier, P.; Pédron, J.; Taghouti, G.; Fischer-Le Saux, M.; Caullireau, E.; Bertrand, C.; Laurent, A.; Chawki, K.; Oulgazi, S.; Moumni, M.; et al. Elevation of Pectobacterium carotovorum subsp. odoriferum to species level as Pectobacterium odoriferum sp. nov., proposal of Pectobacterium brasiliense sp. nov. and Pectobacterium actinidiae sp. nov., emended description of Pectobacterium carotovorum and description of Pectobacterium versatile sp. nov., isolated from streams and symptoms on diverse plants. Int. J. Syst. Evol. Microbiol. 2019, 69, 3207–3216. [Google Scholar] [CrossRef]
- Voronina, M.V.; Kabanova, A.P.; Shneider, M.M.; Korzhenkov, A.A.; Toschakov, S.V.; Miroshnikov, K.K.; Miroshnikov, K.A.; Ignatov, A.N. First Report of Pectobacterium carotovorum subsp. brasiliense Causing Blackleg and Stem Rot Disease of Potato in Russia. Plant. Dis. 2019, 103, 364. [Google Scholar] [CrossRef]
- Malko, A.; Frantsuzov, P.; Nikitin, M.; Statsyuk, N.; Dzhavakhiya, V.; Golikov, A.; Malko, A.; Frantsuzov, P.; Nikitin, M.; Statsyuk, N.; et al. Potato Pathogens in Russia’s Regions: An Instrumental Survey with the Use of Real-Time PCR/RT-PCR in Matrix Format. Pathogens 2019, 8, 18. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, J.B.; Lim, J.A.; Han, S.W.; Heu, S. Genetic diversity of Pectobacterium carotovorum subsp. brasiliensis isolated in Korea. Plant. Pathol. J. 2014, 30, 117–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, X.; Ma, Y.; Liang, S.; Tian, Y.; Yin, S.; Xie, S.; Xie, H. Comparative genomics of 84 Pectobacterium genomes reveals the variations related to a pathogenic lifestyle. BMC Genom. 2018, 19, 889. [Google Scholar] [CrossRef]
- Lukianova, A.A.; Shneider, M.M.; Evseev, P.V.; Shpirt, A.M.; Bugaeva, E.N.; Kabanova, A.P.; Obraztsova, E.A.; Miroshnikov, K.K.; Senchenkova, S.N.; Shashkov, A.S.; et al. Morphologically Different Pectobacterium brasiliense Bacteriophages PP99 and PP101: Deacetylation of O-Polysaccharide by the Tail Spike Protein of Phage PP99 Accompanies the Infection. Front. Microbiol. 2020, 10, 3147. [Google Scholar] [CrossRef] [PubMed]
- Miedzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawełczyk, Z.; Rogóz, P.; Kłak, M.; Wojtasik, E.; et al. Clinical Aspects of Phage Therapy. Adv. Virus Res. 2012, 83, 73–121. [Google Scholar] [CrossRef]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of viral taxonomy: Genetic exchange and failings of phenetic approaches. J. Bacteriol. 2002, 184, 4891–4905. [Google Scholar] [CrossRef]
- Hillyar, C.R.T. Genetic recombination in bacteriophage lambda. Biosci. Horiz. 2012, 5. [Google Scholar] [CrossRef]
- Dragos, A.; B, P.; Hasan, Z.; Lenz-Strube, M.; Kempen, P.J.; Maroti, G.; Kaspar, C.; Bischofs, I.B.; Kovacs, A.T. Phage recombination drives evolution of spore-forming Bacilli. bioRxiv 2020. [Google Scholar] [CrossRef]
- Susskind, M.M.; Botstein, D. Molecular genetics of bacteriophage P22. Microbiol. Rev. 1978, 42, 385–413. [Google Scholar] [CrossRef]
- Casjens, S.R.; Thuman-Commike, P.A. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 2011, 411, 393–415. [Google Scholar] [CrossRef]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Duffy, S.; Sanjuán, R. Point mutation rate of bacteriophage ΦX174. Genetics 2009, 183, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.C.; Castro-Nallar, E.; Fisher, J.N.B.; Breakwell, D.P.; Grose, J.H.; Burnett, S.H. Phage cluster relationships identified through single gene analysis. BMC Genom. 2013, 14, 410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, F.; Zhang, R.; Giovannoni, S.J.; Zhang, Z.; Sun, J.; Du, S.; Rensing, C. Pelagiphages in the Podoviridae family integrate into host genomes. Environ. Microbiol. 2019, 21, 1989–2001. [Google Scholar] [CrossRef]
- Labrie, S.J.; Frois-Moniz, K.; Osburne, M.S.; Kelly, L.; Roggensack, S.E.; Sullivan, M.B.; Gearin, G.; Zeng, Q.; Fitzgerald, M.; Henn, M.R.; et al. Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ. Microbiol. 2013, 15, 1356–1376. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, S.; Jiao, N.; Chen, F. Comparative genomic and phylogenomic analyses reveal a conserved core genome shared by estuarine and oceanic cyanopodoviruses. PLoS ONE 2015, 10, e0142962. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the Marine Virosphere Using Metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schneider, T.D. Information theory based T7-like promoter models: Classification of bacteriophages and differential evolution of promoters and their polymerases. Nucleic Acids Res. 2005, 33, 6172–6187. [Google Scholar] [CrossRef]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef] [PubMed]
- Kiro, R.; Molshanski-Mor, S.; Yosef, I.; Milam, S.L.; Erickson, H.P.; Qimron, U. Gene product 0.4 increases bacteriophage T7 competitiveness by inhibiting host cell division. Proc. Natl. Acad. Sci. USA 2013, 110, 19549–19554. [Google Scholar] [CrossRef]
- Tran, N.Q.; Tabor, S.; Amarasiriwardena, C.J.; Kulczyk, A.W.; Richardson, C.C. Characterization of a nucleotide kinase encoded by bacteriophage T7. J. Biol. Chem. 2012, 287, 29468–29478. [Google Scholar] [CrossRef]
- Cuervo, A.; Pulido-Cid, M.; Chagoyen, M.; Arranz, R.; González-García, V.A.; Garcia-Doval, C.; Castón, J.R.; Valpuesta, J.M.; van Raaij, M.J.; Martín-Benito, J.; et al. Structural Characterization of the Bacteriophage T7 Tail Machinery. J. Biol. Chem. 2013, 288, 26290–26299. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, T.; Globus, R.; Molshanski-Mor, S.; Ben-Shem, A.; Yosef, I.; Qimron, U. A continuous evolution system for contracting the host range of bacteriophage T7. Sci. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Prokhorov, N.S.; Riccio, C.; Zdorovenko, E.L.; Shneider, M.M.; Browning, C.; Knirel, Y.A.; Leiman, P.G.; Letarov, A.V. Function of bacteriophage G7C esterase tailspike in host cell adsorption. Mol. Microbiol. 2017, 105, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Cowen, L.; Menke, M.; King, J.; Berger, B. BETAWRAP: Successful prediction of parallel β-helices from primary sequence reveals an association with many microbial pathogens. Proc. Natl. Acad. Sci. USA 2001, 98, 14819–14824. [Google Scholar] [CrossRef]
- Steven, A.C.; Trus, B.L.; Maizel, J.V.; Unser, M.; Parry, D.A.D.; Wall, J.S.; Hainfeld, J.F.; Studier, F.W. Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. J. Mol. Biol. 1988, 200, 351–365. [Google Scholar] [CrossRef]
- Garcia-Doval, C.; Van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef]
- Hallenbeck, P.C.; Vimr, E.R.; Yu, F.; Bassler, B.; Troy, F.A. Purification and properties of a bacteriophage-induced endo-N-acetylneuraminidase specific for poly-alpha-2,8-sialosyl carbohydrate units. J. Biol. Chem. 1987, 262, 3553–3561. [Google Scholar]
- Mølgaard, A.; Kauppinen, S.; Larsen, S. Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure 2000, 8, 373–383. [Google Scholar] [CrossRef]
- Akoh, C.C.; Lee, G.C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.F. GDSL family of serine esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef]
- Wei, Y.; Schottel, J.L.; Derewenda, U.; Swenson, L.; Patkar, S.; Derewenda, Z.S. A novel variant of the catalytic triad in the streptomyces scabies esterase. Nat. Struct. Biol. 1995, 2, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Michalak, L.; La Rosa, S.L.; Leivers, S.; Lindstad, L.J.; Røhr, Å.K.; Aachmann, F.L.; Westereng, B. A pair of esterases from a commensal gut bacterium remove acetylations from all positions on complex β-mannans. Proc. Natl. Acad. Sci. USA 2020, 117, 7122–7130. [Google Scholar] [CrossRef] [PubMed]
- Mörl, M.; Betat, H.; Rammelt, C. TRNA nucleotidyltransferases: Ancient catalysts with an unusual mechanism of polymerization. Cell. Mol. Life Sci. 2010, 67, 1447–1463. [Google Scholar]
- Yue, D.; Maizels, N.; Weiner, A.M. CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: Characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. RNA 1996, 2, 895–908. [Google Scholar] [PubMed]
- Ackermann, H.W. Bacteriophage observations and evolution. Res. Microbiol. 2003, 154, 245–251. [Google Scholar] [CrossRef]
- Morse, J.W.; Deutscher, M.P. A physiological role for tRNA nucleotidyltransferase during bacteriophage infection. Biochem. Biophys. Res. Commun. 1976, 73, 953–959. [Google Scholar] [CrossRef]
- Wellner, K.; Pöhler, M.T.; Betat, H.; Mörl, M. Dual expression of CCA-adding enzyme and RNase T in Escherichia coli generates a distinct cca growth phenotype with diverse applications. Nucleic Acids Res. 2019, 47, 3631–3639. [Google Scholar] [CrossRef]
- Whiteley, A.T.; Eaglesham, J.B.; de Oliveira Mann, C.C.; Morehouse, B.R.; Lowey, B.; Nieminen, E.A.; Danilchanka, O.; King, D.S.; Lee, A.S.Y.; Mekalanos, J.J.; et al. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 2019, 567, 194–199. [Google Scholar] [CrossRef]
- Ye, Q.; Lau, R.K.; Mathews, I.T.; Birkholz, E.A.; Watrous, J.D.; Azimi, C.S.; Pogliano, J.; Jain, M.; Corbett, K.D. HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol. Cell 2020, 77, 709–722. [Google Scholar] [CrossRef]
- Proux, C.; Van Sinderen, D.; Suarez, J.; Garcia, P.; Ladero, V.; Fitzgerald, G.F.; Desiere, F.; Brüssow, H. The dilemma of phage taxonomy illustrated by comparative genomics of Sfi21-like Siphoviridae in lactic acid bacteria. J. Bacteriol. 2002, 184, 6026–6036. [Google Scholar] [CrossRef]
- Villa, T.G.; Feijoo-Siota, L.; Rama, J.R.; Sánchez-Pérez, A.; Viñas, M. Horizontal Gene Transfer Between Bacteriophages and Bacteria: Antibiotic Resistances and Toxin Production. In Horizontal Gene Transfer; Springer International Publishing: Cham, Switzerland, 2019; pp. 97–142. [Google Scholar]
- Wang, G.H.; Sun, B.F.; Xiong, T.L.; Wang, Y.K.; Murfin, K.E.; Xiao, J.H.; Da Huang, W. Bacteriophage WO can mediate horizontal gene transfer in endosymbiotic Wolbachia genomes. Front. Microbiol. 2016, 7, 1867. [Google Scholar] [CrossRef]
- Frazão, N.; Sousa, A.; Lässig, M.; Gordo, I. Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc. Natl. Acad. Sci. USA 2019, 116, 17906–17915. [Google Scholar] [CrossRef]
- Varble, A.; Meaden, S.; Barrangou, R.; Westra, E.R.; Marraffini, L.A. Recombination between phages and CRISPR−cas loci facilitates horizontal gene transfer in staphylococci. Nat. Microbiol. 2019, 4, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Eggers, C.H.; Gray, C.M.; Preisig, A.M.; Glenn, D.M.; Pereira, J.; Ayers, R.W.; Alshahrani, M.; Acabbo, C.; Becker, M.R.; Bruenn, K.N.; et al. Phage-mediated horizontal gene transfer of both prophage and heterologous DNA by φBB-1, a bacteriophage of Borrelia burgdorferi. Pathog. Dis. 2016, 74, ftw107. [Google Scholar] [CrossRef]
- Heller, K.J. Identification of the phage gene for host receptor specificity by analyzing hybrid phages of T5 and BF23. Virology 1984, 139, 11–21. [Google Scholar] [CrossRef]
- Duplessis, M.; Moineau, S. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol. Microbiol. 2001, 41, 325–336. [Google Scholar] [CrossRef]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol. Lett. 2009, 295, 211–217. [Google Scholar] [CrossRef]
- Schwarzer, D.; Buettner, F.F.R.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Muhlenhoff, M.; et al. A Multivalent Adsorption Apparatus Explains the Broad Host Range of Phage phi92: A Comprehensive Genomic and Structural Analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef]
- Le, S.; He, X.; Tan, Y.; Huang, G.; Zhang, L.; Lux, R.; Shi, W.; Hu, F. Mapping the Tail Fiber as the Receptor Binding Protein Responsible for Differential Host Specificity of Pseudomonas aeruginosa Bacteriophages PaP1 and JG004. PLoS ONE 2013, 8, e68562. [Google Scholar] [CrossRef] [PubMed]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.V.; Lukianova, A.A.; Shneider, M.M.; Korzhenkov, A.A.; Bugaeva, E.N.; Kabanova, A.P.; Miroshnikov, K.K.; Kulikov, E.E.; Toshchakov, S.V.; Ignatov, A.N.; et al. Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches. Microorganisms 2020, 8, 1707. https://doi.org/10.3390/microorganisms8111707
Evseev PV, Lukianova AA, Shneider MM, Korzhenkov AA, Bugaeva EN, Kabanova AP, Miroshnikov KK, Kulikov EE, Toshchakov SV, Ignatov AN, et al. Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches. Microorganisms. 2020; 8(11):1707. https://doi.org/10.3390/microorganisms8111707
Chicago/Turabian StyleEvseev, Peter V., Anna A. Lukianova, Mikhail M. Shneider, Aleksei A. Korzhenkov, Eugenia N. Bugaeva, Anastasia P. Kabanova, Kirill K. Miroshnikov, Eugene E. Kulikov, Stepan V. Toshchakov, Alexander N. Ignatov, and et al. 2020. "Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches" Microorganisms 8, no. 11: 1707. https://doi.org/10.3390/microorganisms8111707
APA StyleEvseev, P. V., Lukianova, A. A., Shneider, M. M., Korzhenkov, A. A., Bugaeva, E. N., Kabanova, A. P., Miroshnikov, K. K., Kulikov, E. E., Toshchakov, S. V., Ignatov, A. N., & Miroshnikov, K. A. (2020). Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches. Microorganisms, 8(11), 1707. https://doi.org/10.3390/microorganisms8111707