Interrelationships of Fiber-Associated Anaerobic Fungi and Bacterial Communities in the Rumen of Bloated Cattle Grazing Alfalfa

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Design, Animal Management, and Assessment of Bloat Scores
2.3. Rumen Sample Collection and Processing
2.4. DNA Extraction and Quality
2.5. Library Construction and Illumina Sequencing
2.6. Bioinformatics
2.7. Statistical Analysis
3. Results
3.1. Bloat Incidence
3.2. Sequencing Results and Phylogenetic Diversity of the Fiber-Associated Microbial Communities
3.3. Diversity of Anaerobic Rumen Fungi as Impacted by Diet and Bloat-Status
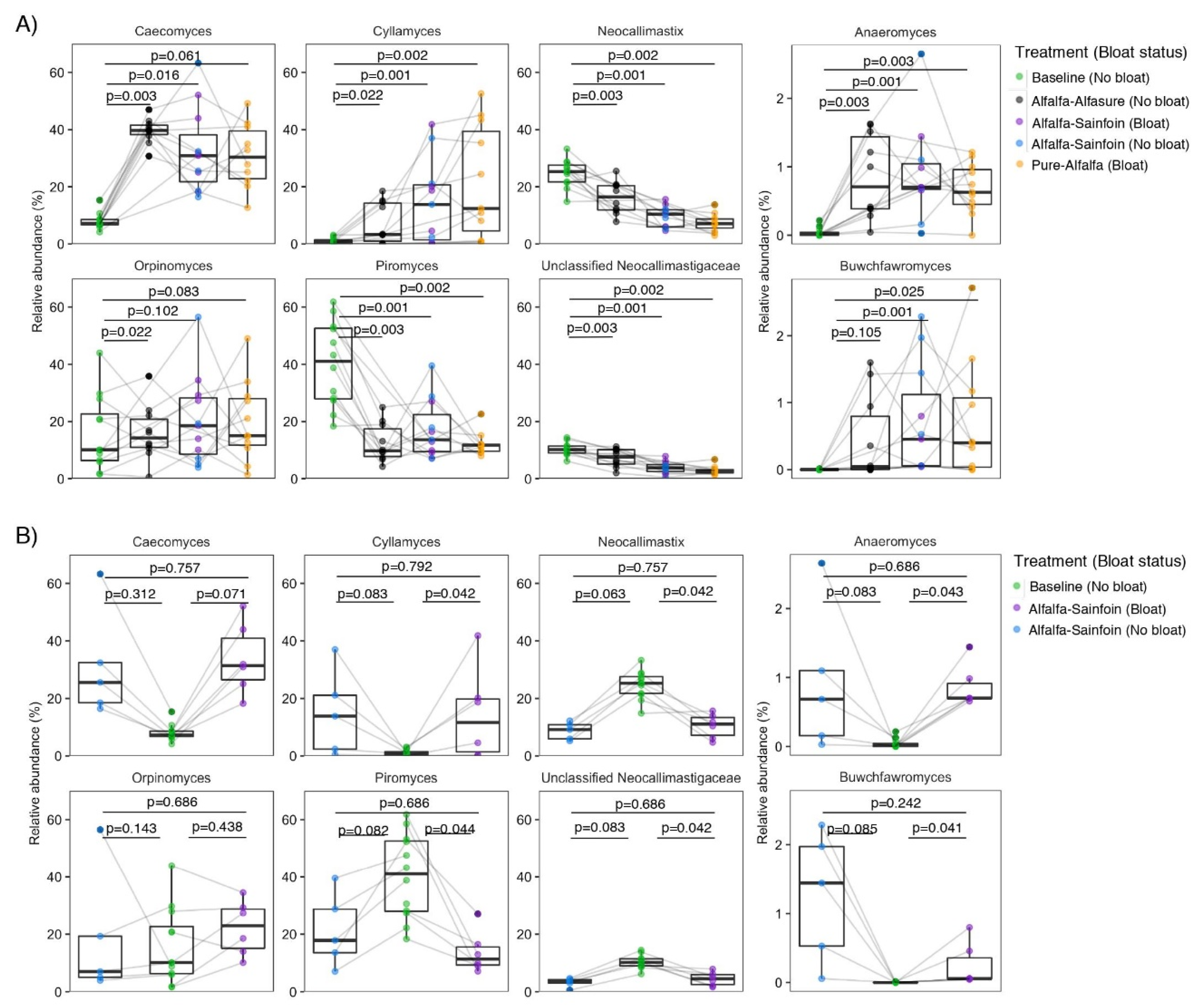
3.4. Composition of Anaerobic Rumen Fungi
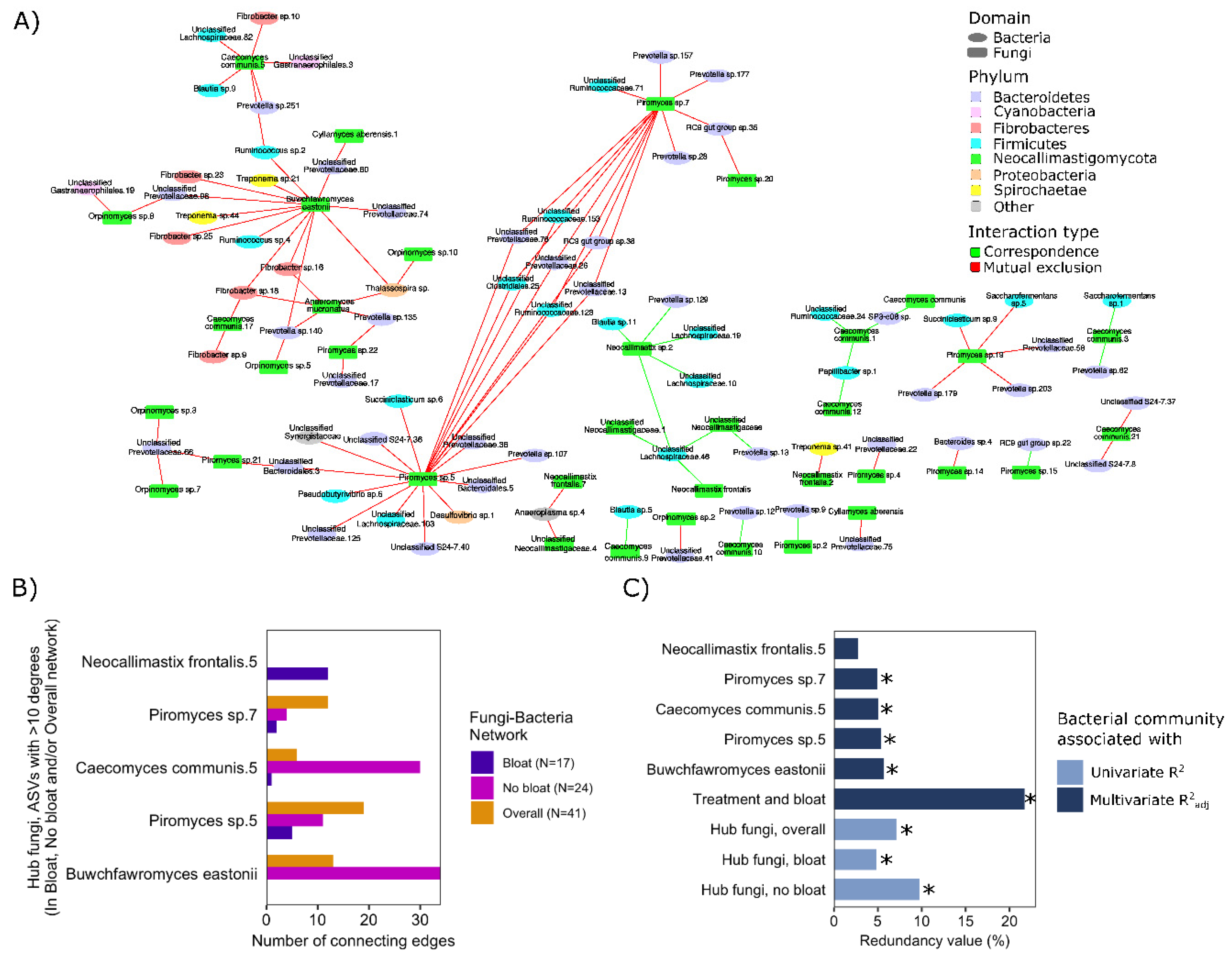
3.5. Co-Occurrence Patterns of Anaerobic Rumen Fungi with the Rumen Bacterial Community in Relation to Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Alfalfa pasture with Alfasure® |
| ARF | Anaerobic rumen fungi |
| AS | Alfalfa—sainfoin |
| ASV | Amplicon sequence variants |
| B | Bloat |
| DM | Dry matter |
| ITS | Internal transcribed spacer |
| NB | no bloat |
| PA | Pure alfalfa pasture |
| rRNA | Ribosomal RNA |
| SRA | Sequence Read Archive |
References
- Popp, J.; McCaughey, W.P.; Cohen, R.D.H.; McAllister, T.A.; Majak, W. Enhancing pasture productivity with alfalfa: A review. Can. J. Plant Sci. 2000, 80, 513–519. [Google Scholar] [CrossRef]
- Wang, Y.; Majak, W.; McAllister, T.A. Frothy bloat in ruminants: Cause, occurrence, and mitigation strategies. Anim. Feed Sci. Technol. 2012, 172, 103–114. [Google Scholar] [CrossRef]
- Nagaraja, T. Rumenology; Springer: Berlin/Heidelberg, Germany, 2016; pp. 39–61. [Google Scholar]
- Majak, W.; McAllister, T.A.; McCartney, D.; Stanford, K.; Cheng, K.-J. Bloat in Cattle; Alberta Agriculture Food and Rural Development Information Packaging Centre: Edmonton, AB, Canada, 2003; pp. 1–24.
- Coulman, B.; Goplen, B.; Makak, W.; McAllister, T.; Cheng, K.-J.; Berg, B.; Hall, J.; McCartney, D.; Acharya, S. A review of the development of a bloat-reduced alfalfa cultivar. Can. J. Plant Sci. 2000, 80, 487–491. [Google Scholar] [CrossRef]
- Majak, W.; Garland, G.J.; Lysyk, T.J.; Olson, M.E. Efficacy of water-soluble feed supplements for the prevention of bloat in cattle. Can. J. Anim. Sci. 2004, 84, 155–157. [Google Scholar] [CrossRef][Green Version]
- McMahon, L.; McAllister, T.A.; Berg, B.P.; Majak, W.; Acharya, S.N.; Popp, J.D.; Coulman, B.E.; Wang, Y.; Cheng, K.-J. A review of the effects of forage condensed tannins on ruminal fermentation and bloat in grazing cattle. Can. J. Plant Sci. 2000, 80, 469–485. [Google Scholar] [CrossRef]
- Azad, E.; Derakhshani, H.; Forster, R.J.; Gruninger, R.J.; Acharya, S.; McAllister, T.A.; Khafipour, E. Characterization of the rumen and fecal microbiome in bloated and non-bloated cattle grazing alfalfa pastures and subjected to bloat prevention strategies. Sci. Rep. 2019, 9, 4272. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Borneman, W. Role of rumen fungi in fiber degradation. J. Dairy Sci. 1990, 73, 3023–3032. [Google Scholar] [CrossRef]
- McAllister, T.A.; Bae, H.D.; Jones, G.A.; Cheng, K.-J. Microbial attachment and feed digestion in the rumen. J. Anim. Sci. 1994, 72, 3004–3018. [Google Scholar] [CrossRef]
- Gordon, G.L.; Phillips, M.W. The role of anaerobic gut fungi in ruminants. Nutr. Res. Rev. 1998, 11, 133–168. [Google Scholar] [CrossRef]
- Srinivasan, K.; Murakami, M.; Nakashimada, Y.; Nishio, N. Efficient production of cellulolytic and xylanolytic enzymes by the rumen anaerobic fungus, Neocallimastix frontalis, in a repeated batch culture. J. Biosci. Bioeng. 2001, 91, 153–158. [Google Scholar] [CrossRef]
- Marvin-Sikkema, F.; Richardson, A.J.; Stewart, C.S.; Gottschal, J.C.; Prins, R.A. Influence of hydrogen-consuming bacteria on cellulose degradation by anaerobic fungi. Appl. Environ. Microbiol. 1990, 56, 3793–3797. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Withers, S.; Joblin, K. Xylanolysis by cocultures of the rumen fungus Neocallimastix frontalis and ruminal bacteria. Lett. Appl. Microbiol. 1991, 12, 232–235. [Google Scholar] [CrossRef]
- Dehority, B.A. Effects of microbial synergism on fibre digestion in the rumen. Proc. Nutr. Soc. 1991, 50, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Lanejekar, V.B.; Dhakephalkar, P.K.; Callaghan, T.M.; Dagar, S.S.; Griffith, G.W.; Elshahed, M.S.; Youssef, N.H. Seven new Neocallimastigomycota genera from wild, zoo-housed, and domesticated herbivores greatly expand the taxonomic diversity of the phylum. Mycologia 2020, 1–28. [Google Scholar] [CrossRef]
- Wang, H.; Li, P.; Liu, X.; Zhang, C.; Lu, Q.; Xi, D.; Yang, R.; Wang, S.; Bai, W.; Yang, Z.; et al. The composition of fungal communities in the rumen of gayals (Bos frontalis), yaks (Bos grunniens), and Yunnan and Tibetan yellow cattle (Bos taurs). Pol. J. Microbiol. 2019, 68, 505. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Elshahed, M.S.; Liggenstoffer, A.S.; Griffith, G.W.; Youssef, N.H. Pecoramyces ruminantium, gen. nov. snov. an anaerobic gut fungus from the feces of cattle and sheep. Mycologia 2017, 109, 231–243. [Google Scholar] [CrossRef]
- Kim, M.; Park, T.; Yu, Z. Metagenomic investigation of gastrointestinal microbiome in cattle. Asian-Aust. J. Anim. Sci. 2017, 30, 1515–1528. [Google Scholar] [CrossRef]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef]
- Callaghan, T.M.; Podmirseg, S.M.; Hohlweck, D.; Edwards, J.E.; Puniya, A.K.; Dagar, S.S.; Griffith, G.W. Buwchfawromyces eastonii gen. nov. snov.: A new anaerobic fungus (Neocallimastigomycota) isolated from buffalo faeces. MycoKeys 2015, 9, 11–28. [Google Scholar] [CrossRef]
- Edwards, J.E.; Forster, R.J.; Callaghan, T.M.; Dollhofer, V.; Dagar, S.S.; Cheng, Y.; Chang, J.; Kittelmann, S.; Fliegerova, K.; Puniya, A.K.; et al. PCR and omics based techniques to study the diversity, ecology and biology of anaerobic fungi: Insights, challenges and opportunities. Front. Microbiol. 2017, 8, 1657. [Google Scholar] [CrossRef] [PubMed]
- Gruninger, R.J.; Puniya, A.K.; Callaghan, T.M.; Edwards, J.E.; Youssef, N.; Dagar, S.S.; Fliegerova, K.; Griffith, G.W.; Forster, R.; Tsang, A.; et al. Anaerobic fungi (phylum Neocallimastigomycota): Advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol. Ecol. 2014, 90, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Canadian Council on Animal Care. CCAC Guidelines on: The Care and Use of Farm. Animals in Research, Teaching and Testing; Canadian Council on Animal Care: Ottawa, ON, Canada, 2009. [Google Scholar]
- Majak, W.; Hall, J.; Howarth, R. The distribution of chlorophyll in rumen contents and the onset of bloat in cattle. Can. J. Anim. Sci. 1986, 66, 97–102. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems 2016, 1, e00009-15. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 23955772. [Google Scholar] [CrossRef]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016, 081257. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Tedersoo, L.; Ryberg, M.; Kristiansson, E.; Hartmann, M.; Unterseher, M.; Porter, T.M.; Bengstsson-Palme, J.; Walker, D.M.; de Sousa, F.; et al. A comprehensive, automatically updated fungal ITS sequence dataset for reference-based chimera control in environmental sequencing efforts. Microbes Environ. 2015, 30, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5-6; 2019. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 1 September 2019).
- Gloor, B.G.; Reid, G. Compositional analysis: A valid approach to analyze microbiome high-throughput sequencing data. Can. J. Microbiol. 2016, 26, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Palarea-Albaladejo, J.; Martin-Fernandez, J.A. zCompositions. R package for multivariate imputation of left-censored data under a compositional approach. Chemom. Intell. Lab. Syst. 2015, 143, 85–96. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. CoNet app: Inference of Biological Association Networks Using Cytoscape. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Brown, M.B. 400: A method for combining non-independent, one-sided tests of significance. Biometrics 1975, 31, 987–992. [Google Scholar] [CrossRef]
- Wheeler, B.; Torchiano, M. lmPerm: Permutation Tests for Linear Models, R Package Version 2.1.0; 2016. Available online: https://CRAN.R-project.org/package=lmPerm (accessed on 2 August 2016).
- Zhang, Y.; Li, F.; Chen, Y.; Wu, H.; Meng, Q.; Guan, L.L. Metatranscriptomic profiling reveals the effect of breed on active rumen eukaryotic composition in beef cattle with varied feed efficiency. Front. Microbiol. 2020, 11, 367. [Google Scholar] [CrossRef]
- Kittelmann, S.; Seedorf, H.; Walters, W.A.; Clemente, J.C.; Knight, R.; Gordon, J.I.; Janssen, P.H. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE 2013, 8, e47879. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Liggenstoffer, A.S.; Youssef, N.; Couger, M.B.; Elshahed, M.S. Phylogenetic diversity and community structure of anaerobic gut fungi (phylum Neocallimastigomycota) in ruminant and non-ruminant herbivores. ISME J. 2010, 4, 1225–1235. [Google Scholar] [CrossRef]
- Griffith, G.W.; Baker, S.; Fliegerova, K.; Liggenstroffer, A.; van der Giezen, M.; Voigt, K.; Beaks, G. Anaerobic fungi: Neocallimastigomycota. IMA Fungus 2010, 1, 181–185. [Google Scholar] [CrossRef]
- Fliegerova, K.; Kaerger, K.; Kirk, P.; Voigt, K. Rumen Fungi. In Rumen Microbiology: From Evolution to Revolution; Puniya, A.K., Singh, R., Kamra, D.N., Eds.; Springer: New Delhi, India, 2015; pp. 97–112. [Google Scholar]
- Mizrahi, I. The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2013; pp. 533–544. [Google Scholar]
- Lee, S.S.; Choi, C.K.; Ahn, B.H.; Moon, Y.H.; Kim, C.H.; Ha, J.K. In vitro stimulation of rumen microbial fermentation by a rumen anaerobic fungal culture. Anim. Feed Sci. Technol. 2004, 115, 215–226. [Google Scholar] [CrossRef]
- Klevenhusen, F.; Petri, R.M.; Kleefisch, M.-T.; Khiaosa-Ard, R.; Metzler-Zebeli, B.U.; Zebeli, Q. Changes in fibre-adherent and fluid-associated microbial communities and fermentation profiles in the rumen of cattle fed diets differing in hay quality and concentrate amount. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Bekele, A.Z.; Koike, S.; Kobayashi, Y. Phylogenetic diversity and dietary association of rumen Treponema revealed using group-specific 16S rRNA gene-based analysis. FEMS Microbiol. Lett. 2011, 316, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.S.; Duncan, S.H.; Richardson, A.J.; Blackwell, C.; Begbie, R. The inhibition of fungal cellulolysis by cell-free preparations from ruminococci. FEMS Microbiol. Lett. 1992, 97, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Dehority, B.A. Rumen Microbiology; Nottingham University Press: Nottingham, UK, 2003. [Google Scholar]
- Liu, J.; Pu, Y.-Y.; Xie, Q.; Wang, J.-K.; Liu, J.X. Pectin induces an in vitro rumen microbial population shift attributed to the pectinolytic Treponema group. Curr. Microbiol. 2015, 70, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, J.-K.; Zhu, W.; Pu, Y.-Y.; Guan, L.-L.; Liu, J.-X. Monitoring the rumen pectinolytic bacteria Treponema saccharophilum using real-time PCR. FEMS Microbiol. Ecol. 2014, 87, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Bento, M.H.L.; Acamovic, T.; Makkar, H.P.S. The influence of tannin, pectin and polyethylene glycol on attachment of 15N-labelled rumen microorganisms to cellulose. Anim. Feed Sci. Technol. 2005, 122, 41–57. [Google Scholar] [CrossRef]
- McSweeney, C.S.; Palmer, B.; Kennedy, B.; Krause, D. Effect of calliandra tannins on rumen microbial function. Anim. Prod. Aust. 1998, 22, 289. [Google Scholar] [CrossRef]
- McSweeney, C.S.; Palmer, B.; McNeill, D.M.; Krause, D.O. Microbial interactions with tannins: Nutritional consequences for ruminants. Anim. Feed Sci. Technol. 2001, 91, 83–93. [Google Scholar] [CrossRef]
- Sottie, E.T.; Acharya, S.N.; McAllister, T.A.; Thomas, J.E.; Wang, Y.; Iwaasa, A.D. Alfalfa pasture bloat can be eliminated by intermixing with newly-developed sainfoin population. Agron. J. 2013, 106, 1470–1478. [Google Scholar] [CrossRef]
- McAllister, T.A.; Bae, H.D.; Yanke, L.J.; Cheng, K.-J.; Muir, A. Effect of condensed tannins from birdsfoot trefoil on endoglucanase activity and the digestion of cellulose filter paper by ruminal fungi. Can. J. Microbiol. 1994, 40, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Makkar, H.P.S. In vitro gas methods for evaluation of feeds containing phytochemicals. Anim. Feed Sci. Technol. 2005, 123–124, 291–302. [Google Scholar] [CrossRef]
- Stenuit, B.; Agathos, S.N. Deciphering microbial community robustness through synthetic ecology and molecular systems synecology. Curr. Opin. Biotechnol. 2015, 33, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, I. Rumen Symbioses, in The Prokaryotes: Prokaryotic Biology and Symbiotic Associations; Rosenberg, E., Delong, E.F., Thompson, F., Lory, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 533–544. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Period | Treatment 1 | |||||
|---|---|---|---|---|---|---|
| Pure Alfalfa | Alfalfa + Alfasure® | Alfalfa + Sainfoin | ||||
| Bloat Incidence 2 | Steers | Bloat Incidence | Steers | Bloat Incidence | Steers | |
| Period 1 | NB | 0 | NB | 4 | NB | 0 |
| B | 4 | B | 0 | B | 4 | |
| Period 2 | NB | 0 | NB | 3 | NB | 3 |
| B | 3 | B | 0 | B | 1 | |
| Period 3 | NB | 0 | NB | 3 | NB | 2 |
| B | 4 | B | 0 | B | 1 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azad, E.; Fehr, K.B.; Derakhshani, H.; Forster, R.; Acharya, S.; Khafipour, E.; McGeough, E.; McAllister, T.A. Interrelationships of Fiber-Associated Anaerobic Fungi and Bacterial Communities in the Rumen of Bloated Cattle Grazing Alfalfa. Microorganisms 2020, 8, 1543. https://doi.org/10.3390/microorganisms8101543
Azad E, Fehr KB, Derakhshani H, Forster R, Acharya S, Khafipour E, McGeough E, McAllister TA. Interrelationships of Fiber-Associated Anaerobic Fungi and Bacterial Communities in the Rumen of Bloated Cattle Grazing Alfalfa. Microorganisms. 2020; 8(10):1543. https://doi.org/10.3390/microorganisms8101543
Chicago/Turabian StyleAzad, Elnaz, Kelsey B. Fehr, Hooman Derakhshani, Robert Forster, Surya Acharya, Ehsan Khafipour, Emma McGeough, and Tim A. McAllister. 2020. "Interrelationships of Fiber-Associated Anaerobic Fungi and Bacterial Communities in the Rumen of Bloated Cattle Grazing Alfalfa" Microorganisms 8, no. 10: 1543. https://doi.org/10.3390/microorganisms8101543
APA StyleAzad, E., Fehr, K. B., Derakhshani, H., Forster, R., Acharya, S., Khafipour, E., McGeough, E., & McAllister, T. A. (2020). Interrelationships of Fiber-Associated Anaerobic Fungi and Bacterial Communities in the Rumen of Bloated Cattle Grazing Alfalfa. Microorganisms, 8(10), 1543. https://doi.org/10.3390/microorganisms8101543