The Chlamydia trachomatis Extrusion Exit Mechanism Is Regulated by Host Abscission Proteins


Abstract
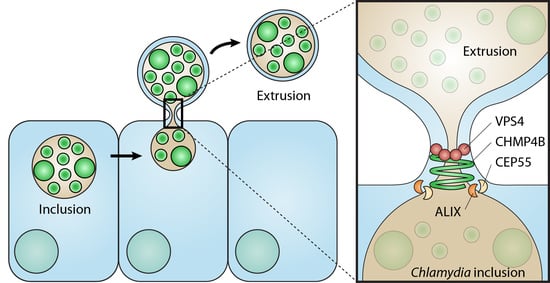
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chlamydia Propagation and Infections
2.2. Extrusion Isolation
2.3. Immunofluorescence and Live Fluorescence Microscopy
2.4. Image Processing and Analysis
2.5. RNA Interference
2.6. Real Time Quantitative PCR
2.7. Cell Multinucleation Assay
2.8. Statistical Analysis
3. Results
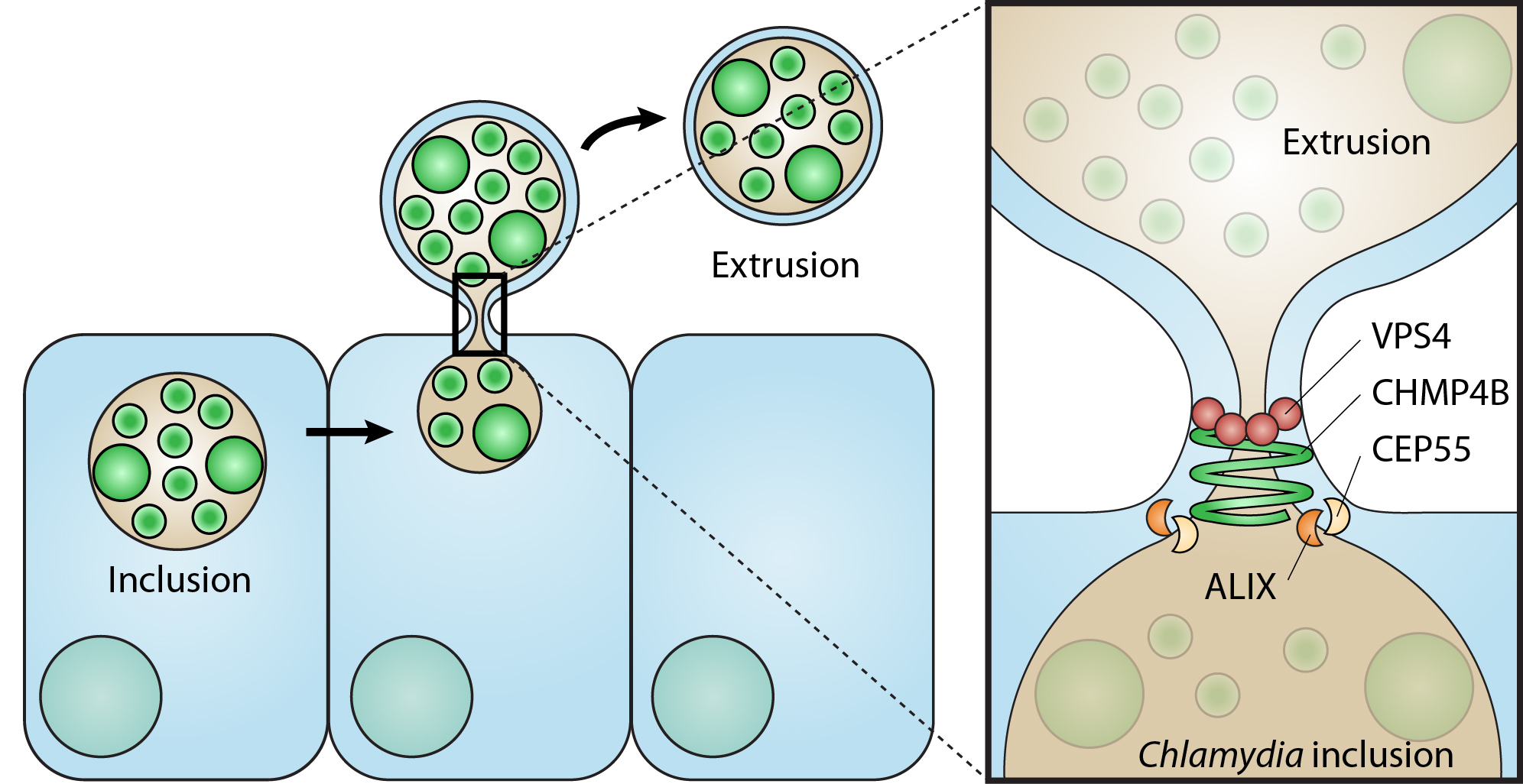
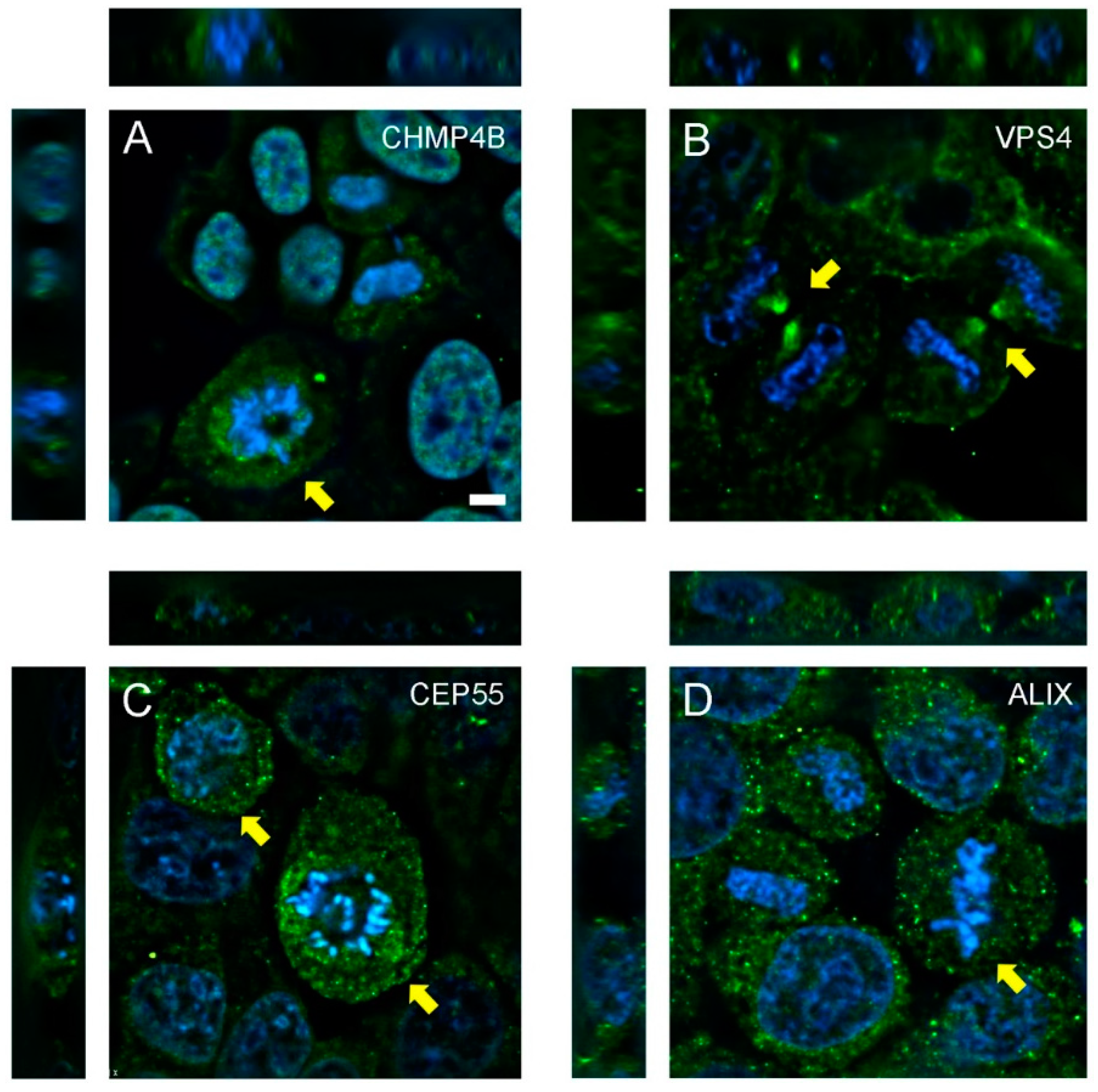
3.1. Abscission Protein Distributions in Dividing HeLa Cells
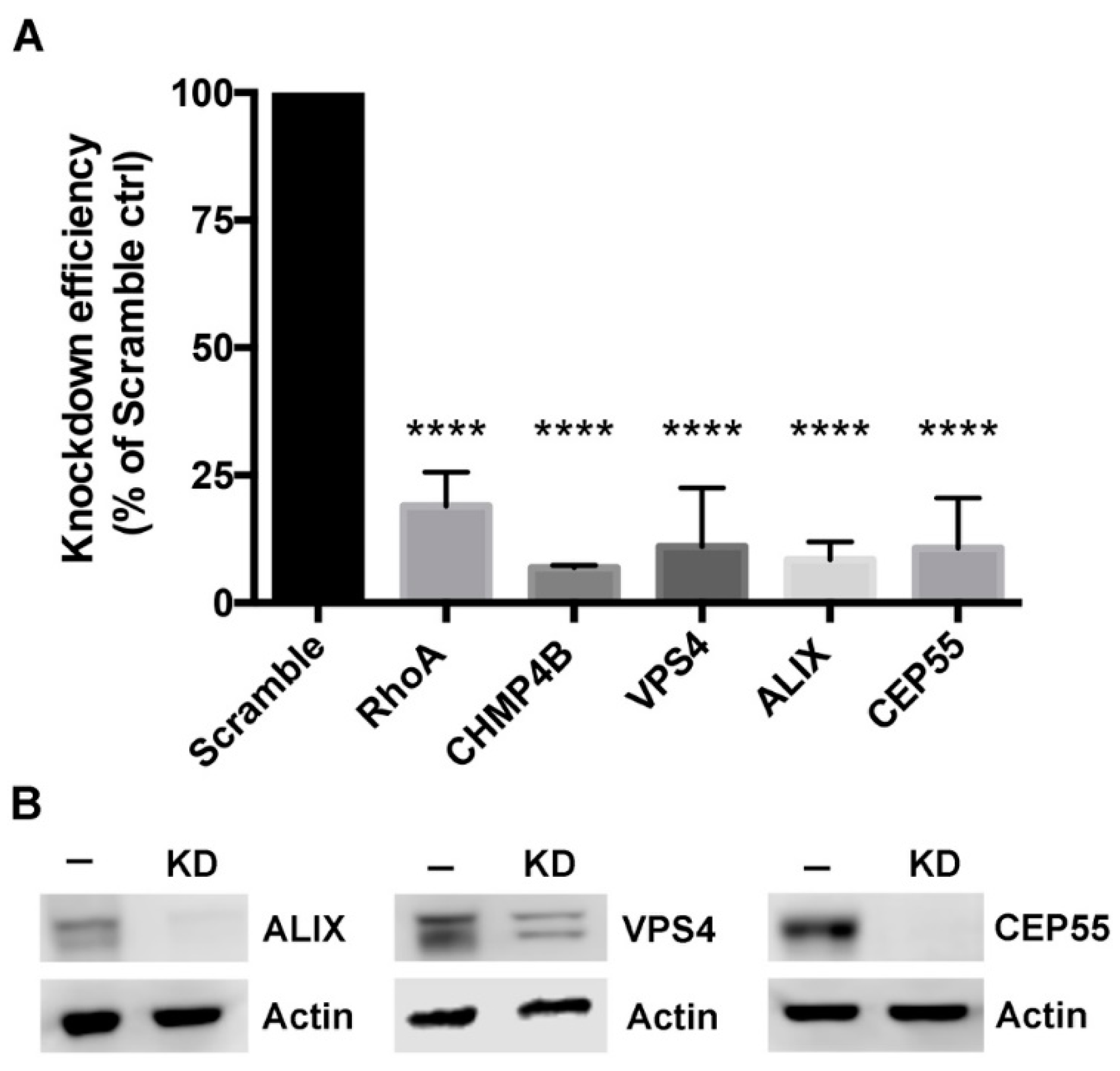
3.2. Depletion of Abscission Proteins in HeLa Cells
3.3. Multinucleated Phenotypes Observed Following siRNA Transfection
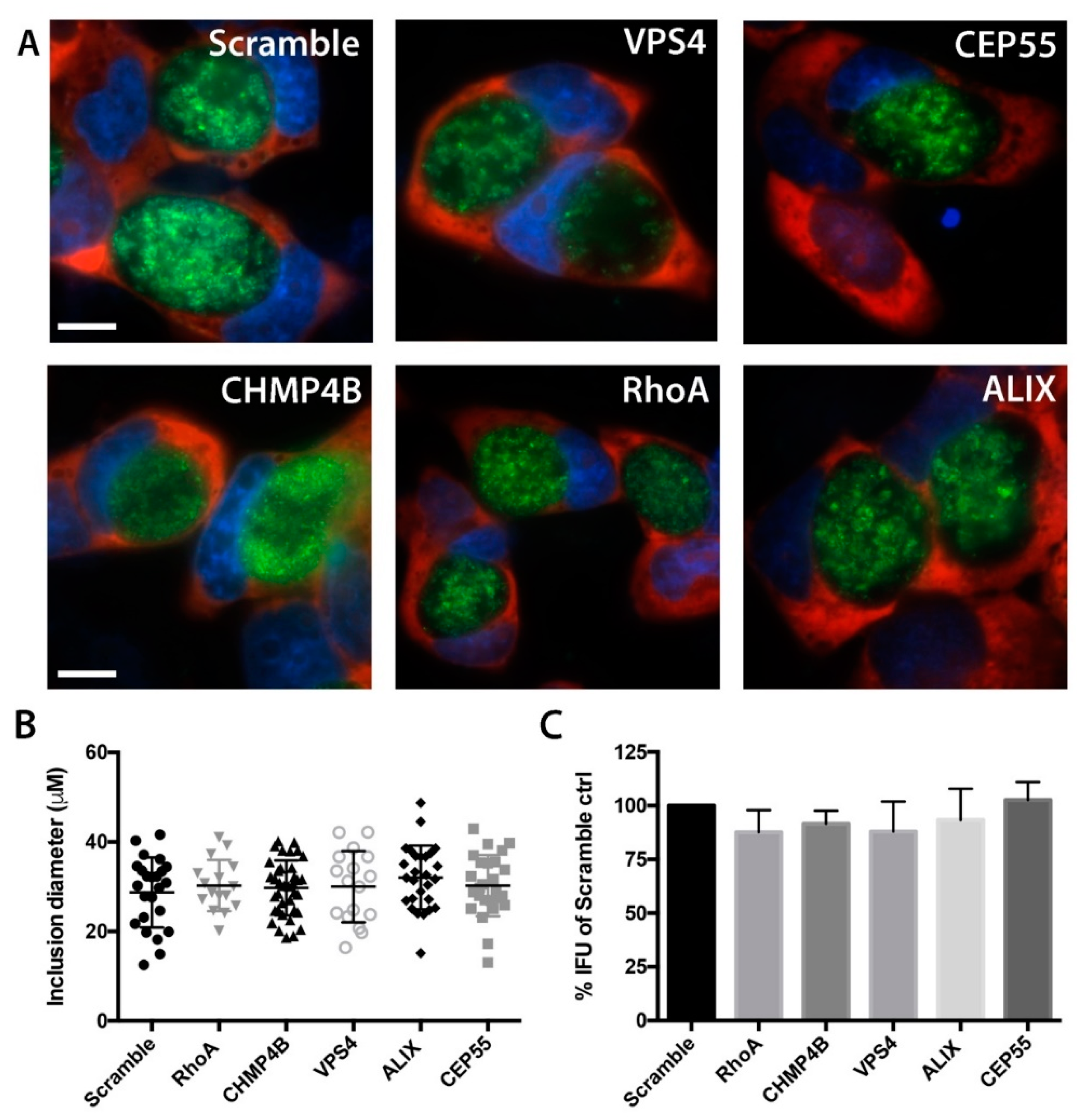
3.4. Chlamydial Inclusion Morphology and Infectivity Not Affected by Depletion of Abscission Proteins
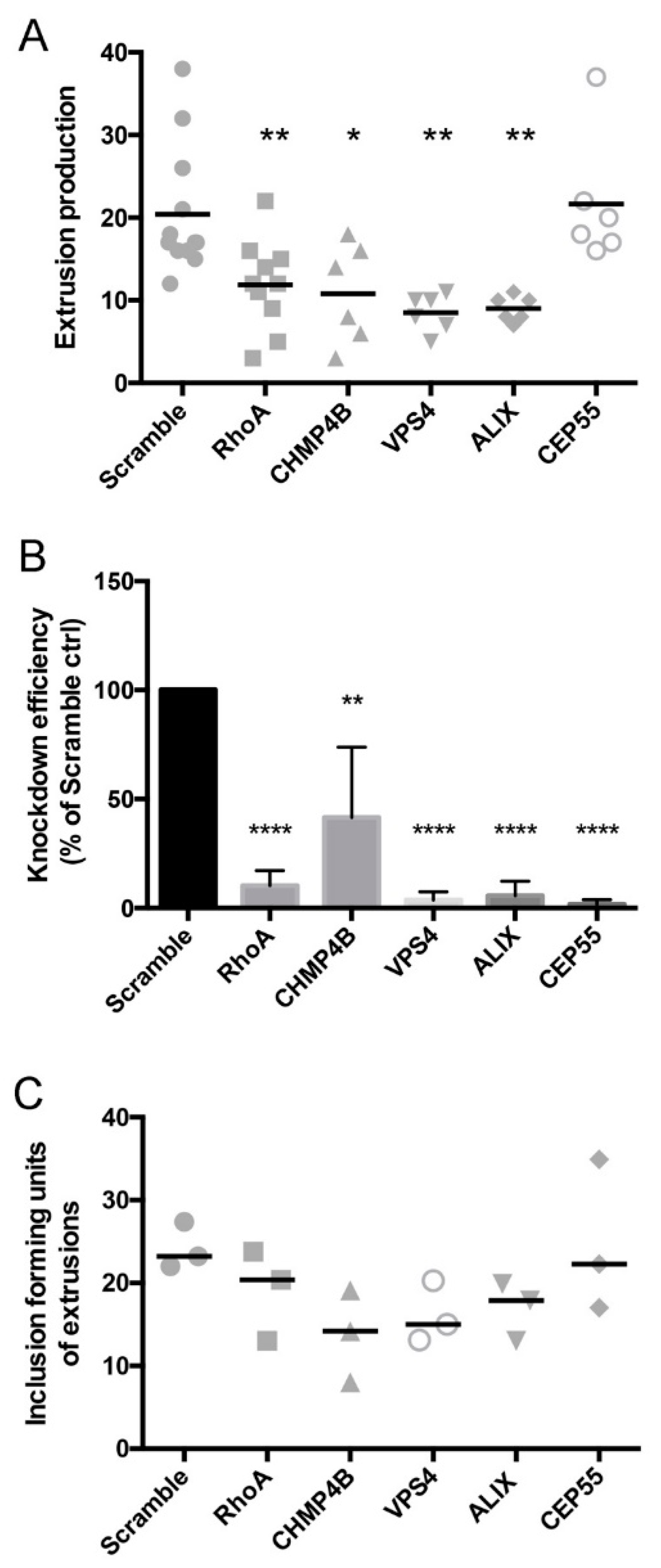
3.5. Depletion of Abscission Proteins Partially Inhibited Extrusion Production
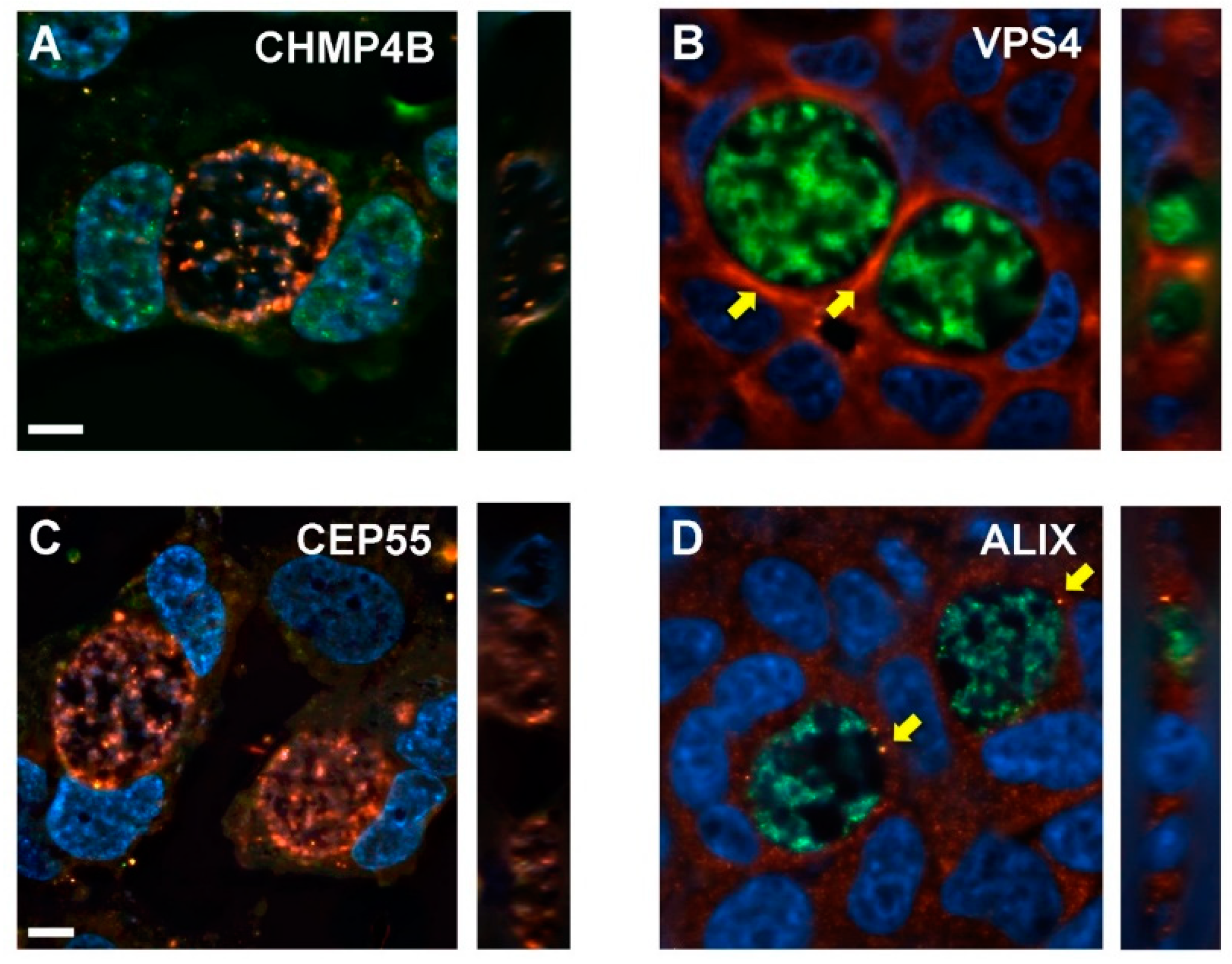
3.6. Distributions of Abscission Proteins on Late-Stage Chlamydial Inclusions
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Brunham, R.C.; Rey-Ladino, J. Immunology of Chlamydia infection: Implications for a Chlamydia trachomatis vaccine. Nat. Rev. Immunol. 2005, 5, 149–161. [Google Scholar] [CrossRef]
- Hafner, L.M.; Wilson, D.P.; Timms, P. Development status and future prospects for a vaccine against Chlamydia trachomatis infection. Vaccine 2014, 32, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Hybiske, K.; Stephens, R.S. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 2007, 104, 11430–11435. [Google Scholar] [CrossRef] [PubMed]
- Zuck, M.; Sherrid, A.; Suchland, R.; Ellis, T.; Hybiske, K. Conservation of extrusion as an exit mechanism for Chlamydia. Pathog. Dis. 2016, 74, ftw093. [Google Scholar]
- Chin, E.; Kirker, K.; Zuck, M.; James, G.; Hybiske, K. Actin recruitment to the Chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors. PLoS ONE 2012, 7, e46949. [Google Scholar]
- Lutter, E.I.; Barger, A.C.; Nair, V.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 2013, 3, 1921–1931. [Google Scholar] [CrossRef]
- Grieshaber, S.S.; Grieshaber, N.A.; Miller, N.; Hackstadt, T. Chlamydia trachomatis causes centrosomal defects resulting in chromosomal segregation abnormalities. Traffic 2006, 7, 940–949. [Google Scholar] [CrossRef]
- Alzhanov, D.T.; Weeks, S.K.; Burnett, J.R.; Rockey, D.D. Cytokinesis is blocked in mammalian cells transfected with Chlamydia trachomatis gene CT223. BMC Microbiol. 2009, 9, 2. [Google Scholar] [CrossRef]
- Greene, W.; Zhong, G. Inhibition of host cell cytokinesis by Chlamydia trachomatis infection. J. Infect. 2003, 47, 45–51. [Google Scholar] [CrossRef]
- Balsara, Z.R.; Misaghi, S.; Lafave, J.N.; Starnbach, M.N. Chlamydia trachomatis infection induces cleavage of the mitotic cyclin B1. Infect. Immun. 2006, 74, 5602–5608. [Google Scholar] [CrossRef]
- Agromayor, M.; Martin-Serrano, J. Knowing when to cut and run: Mechanisms that control cytokinetic abscission. Trends Cell Biol. 2013, 23, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Mierzwa, B.; Gerlich, D.W. Cytokinetic abscission: Molecular mechanisms and temporal control. Dev. Cell 2014, 31, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Colf, L.A.; Karren, M.A.; Sandrin, V.; Rodesch, C.K.; Sundquist, W.I. Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 12889–12894. [Google Scholar] [CrossRef]
- Nähse, V.; Christ, L.; Stenmark, H.; Campsteijn, C. The Abscission Checkpoint: Making It to the Final Cut. Trends Cell Biol. 2017, 27, 1–11. [Google Scholar] [CrossRef]
- Chen, C.-T.; Hehnly, H.; Doxsey, S.J. Orchestrating vesicle transport, ESCRTs and kinase surveillance during abscission. Nat. Rev. Mol. Cell Biol. 2012, 13, 483–488. [Google Scholar] [CrossRef]
- Mueller, M.; Adell, M.A.Y.; Teis, D. Membrane abscission: First glimpse at dynamic ESCRTs. Curr. Biol. 2012, 22, R603–R605. [Google Scholar] [CrossRef][Green Version]
- Adell, M.A.Y.; Teis, D. Assembly and disassembly of the ESCRT-III membrane scission complex. FEBS Lett. 2011, 585, 3191–3196. [Google Scholar] [CrossRef]
- Morita, E.; Sandrin, V.; McCullough, J.; Katsuyama, A.; Baci Hamilton, I.; Sundquist, W.I. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 2011, 9, 235–242. [Google Scholar] [CrossRef]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef]
- Pornillos, O.; Higginson, D.S.; Stray, K.M.; Fisher, R.D.; Garrus, J.E.; Payne, M.; He, G.-P.; Wang, H.E.; Morham, S.G.; Sundquist, W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003, 162, 425–434. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Carpp, L.N.; Galler, R.; Bonaldo, M.C. Interaction between the yellow fever virus nonstructural protein NS3 and the host protein Alix contributes to the release of infectious particles. Microbes Infect. 2011, 13, 85–95. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Colf, L.A.; Sundquist, W.I. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 2013, 82, 663–692. [Google Scholar] [CrossRef]
- Guizetti, J.; Schermelleh, L.; Mäntler, J.; Maar, S.; Poser, I.; Leonhardt, H.; Müller-Reichert, T.; Gerlich, D.W. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science 2011, 331, 1616–1620. [Google Scholar] [CrossRef] [PubMed]
- Elia, N.; Sougrat, R.; Spurlin, T.A.; Hurley, J.H.; Lippincott-Schwartz, J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. USA 2011, 108, 4846–4851. [Google Scholar] [CrossRef]
- Hurley, J.H.; Emr, S.D. The ESCRT complexes: Structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 277–298. [Google Scholar] [CrossRef]
- Carlton, J.G.; Martin-Serrano, J. Parallels between cytokinesis and retroviral budding: A role for the ESCRT machinery. Science 2007, 316, 1908–1912. [Google Scholar] [CrossRef]
- Lipkin, E.S.; Moncada, J.V.; Shafer, M.A.; Wilson, T.E.; Schachter, J. Comparison of monoclonal antibody staining and culture in diagnosing cervical chlamydial infection. J. Clin. Microbiol. 1986, 23, 114–117. [Google Scholar] [PubMed]
- Wang, Y.; Kahane, S.; Cutcliffe, L.T.; Skilton, R.J.; Lambden, P.R.; Clarke, I.N. Development of a transformation system for Chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011, 7, e1002258. [Google Scholar] [CrossRef] [PubMed]
- Zuck, M.; Ellis, T.; Venida, A.; Hybiske, K. Extrusions are phagocytosed and promote Chlamydia survival within macrophages. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef]
- Xiao, H.; Xu, D.; Chen, P.; Zeng, G.; Wang, X.; Zhang, X. Identification of Five Genes as a Potential Biomarker for Predicting Progress and Prognosis in Adrenocortical Carcinoma. J. Cancer 2018, 9, 4484–4495. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, S.C.; Starnbach, M.N. PD-L1 limits the mucosal CD8+ T cell response to Chlamydia trachomatis. J. Immunol. 2014, 192, 1079–1090. [Google Scholar] [CrossRef]
- Robijns, J.; Molenberghs, F.; Sieprath, T.; Corne, T.D.J.; Verschuuren, M.; De Vos, W.H. In silico synchronization reveals regulators of nuclear ruptures in lamin A/C deficient model cells. Sci. Rep. 2016, 6, 30325. [Google Scholar] [CrossRef]
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. ESCRT-III controls nuclear envelope reformation. Nature 2015, 522, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Zhadina, M.; Bieniasz, P.D.; Simon, S.M. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 2011, 13, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Zuck, M.; Feng, C.; Hybiske, K. Using Fluorescent Proteins to Visualize and Quantitate Chlamydia Vacuole Growth Dynamics in Living Cells. J. Vis. Exp. 2015, 104, e51131. [Google Scholar] [CrossRef]
- Morita, E.; Sandrin, V.; Chung, H.-Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227. [Google Scholar] [CrossRef]
- Vromman, F.; Perrinet, S.; Gehre, L.; Subtil, A. The DUF582 Proteins of Chlamydia trachomatis Bind to Components of the ESCRT Machinery, Which Is Dispensable for Bacterial Growth In vitro. Front. Cell. Infect. Microbiol. 2016, 6, 123. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Target Sequence |
|---|---|
| Human CEP55 (55165) | CUGAGUGAAUUUCGAAGAA |
| Non-targeting pool | UGGUUUACAUGUCGACUAA |
| UGGUUUACAUGUUGUGUGA | |
| UGGUUUACAUGUUUUCCUA | |
| Human ALIX | CAGAUCUGCUUGACAUUUA |
| Human VPS4 | CCACAAACAUCCCAUGGGU |
| Human CHMP4B | CCAUCGAGUUCCAGCGGGA |
| AGAAGAGUUUGACGAGGAU | |
| CGGAAGAGAUGUUAAGCAA | |
| UGGAAAGGGUCGACUGGUU |
| Gene Name | Sequence 5’ to 3’ |
|---|---|
| GAPDH fwd | GGTGCTGAGTATGTCGTGGA |
| GAPDH rev | CGGAGATGATGACCCTTTTG |
| ALIX fwd | GACGCTCCTGAGATATTATGATCAG |
| ALIX rev | ACACACAGCTCTTTTCATATCCTAAGC |
| VPS4 fwd | GGAAGACGGAAGGCTACTCG |
| VPS4 rev | AGGGGCCACAGACCTTTTTG |
| CEP55 fwd | GGAGGGCAGACCATTTCAGAG |
| CEP55 rev | AGGCTTCGATCCCCACTTAC |
| RhoA fwd | GTGGATGGAAAGCAGGTAGAG |
| RhoA rev | TAACATCGGTATCTGGGTAGGA |
| CHMP4B fwd | GGAGAAGAGTTTGACGAGGATG |
| CHMP4B rev | CTGTTTCGGGTCCACTGATT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuck, M.; Hybiske, K. The Chlamydia trachomatis Extrusion Exit Mechanism Is Regulated by Host Abscission Proteins. Microorganisms 2019, 7, 149. https://doi.org/10.3390/microorganisms7050149
Zuck M, Hybiske K. The Chlamydia trachomatis Extrusion Exit Mechanism Is Regulated by Host Abscission Proteins. Microorganisms. 2019; 7(5):149. https://doi.org/10.3390/microorganisms7050149
Chicago/Turabian StyleZuck, Meghan, and Kevin Hybiske. 2019. "The Chlamydia trachomatis Extrusion Exit Mechanism Is Regulated by Host Abscission Proteins" Microorganisms 7, no. 5: 149. https://doi.org/10.3390/microorganisms7050149
APA StyleZuck, M., & Hybiske, K. (2019). The Chlamydia trachomatis Extrusion Exit Mechanism Is Regulated by Host Abscission Proteins. Microorganisms, 7(5), 149. https://doi.org/10.3390/microorganisms7050149