Metabolic Modeling of Pectobacterium parmentieri SCC3193 Provides Insights into Metabolic Pathways of Plant Pathogenic Bacteria

 ,
,  ,
, 
 and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Metabolic Network Reconstruction and Model Refinement
2.2. Biomass
2.3. Metabolic Modeling
2.4. Bacterial Strains and Culture Conditions
2.5. Experimental High-Throughput Phenotypic Characterization on P. parmentieri SCC3193
2.6. OmniLog™ Data Processing and Analysis
2.7. In Silico Environmental Representations
2.8. Flux Balance Analysis and Flux Variability Analysis
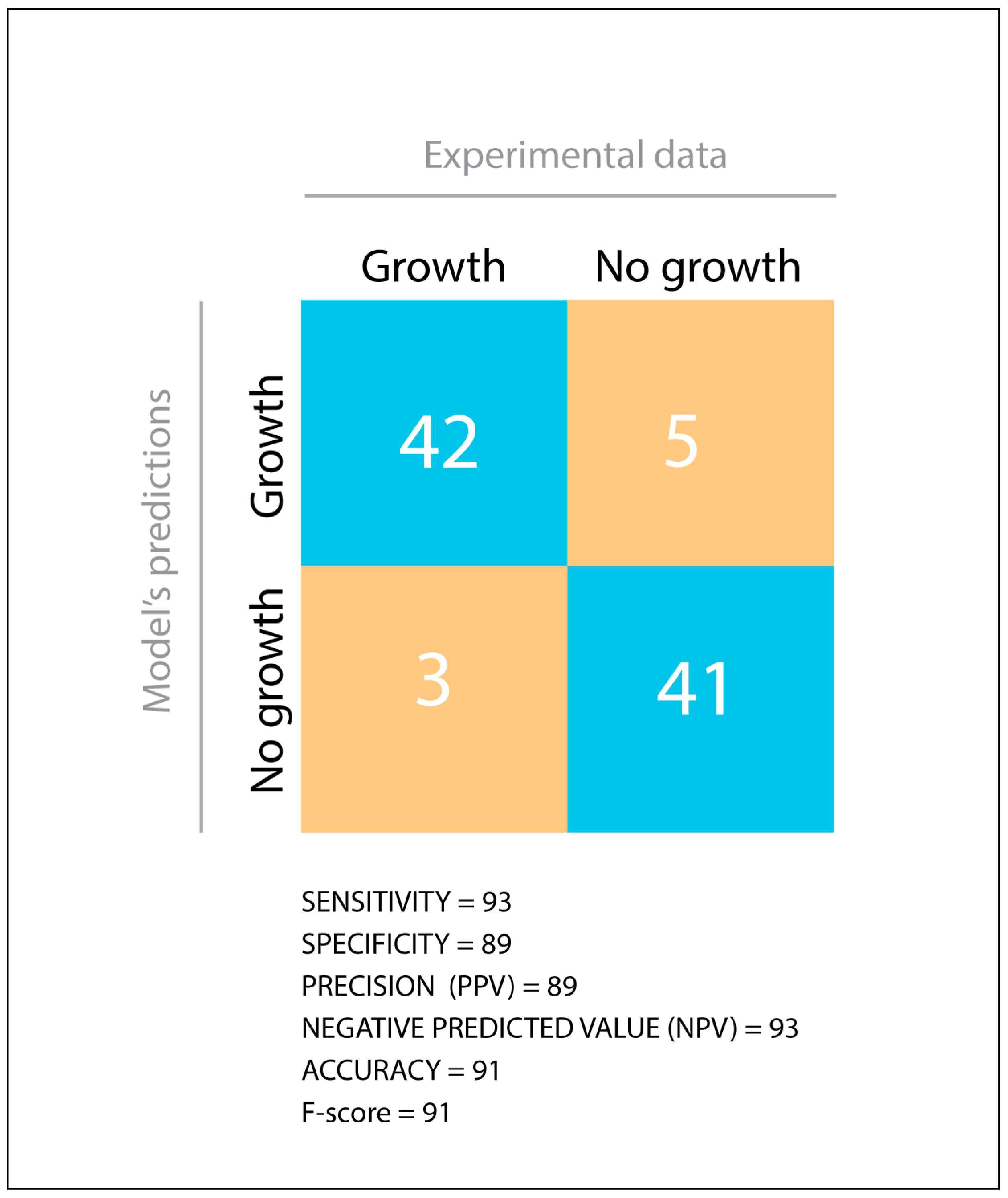
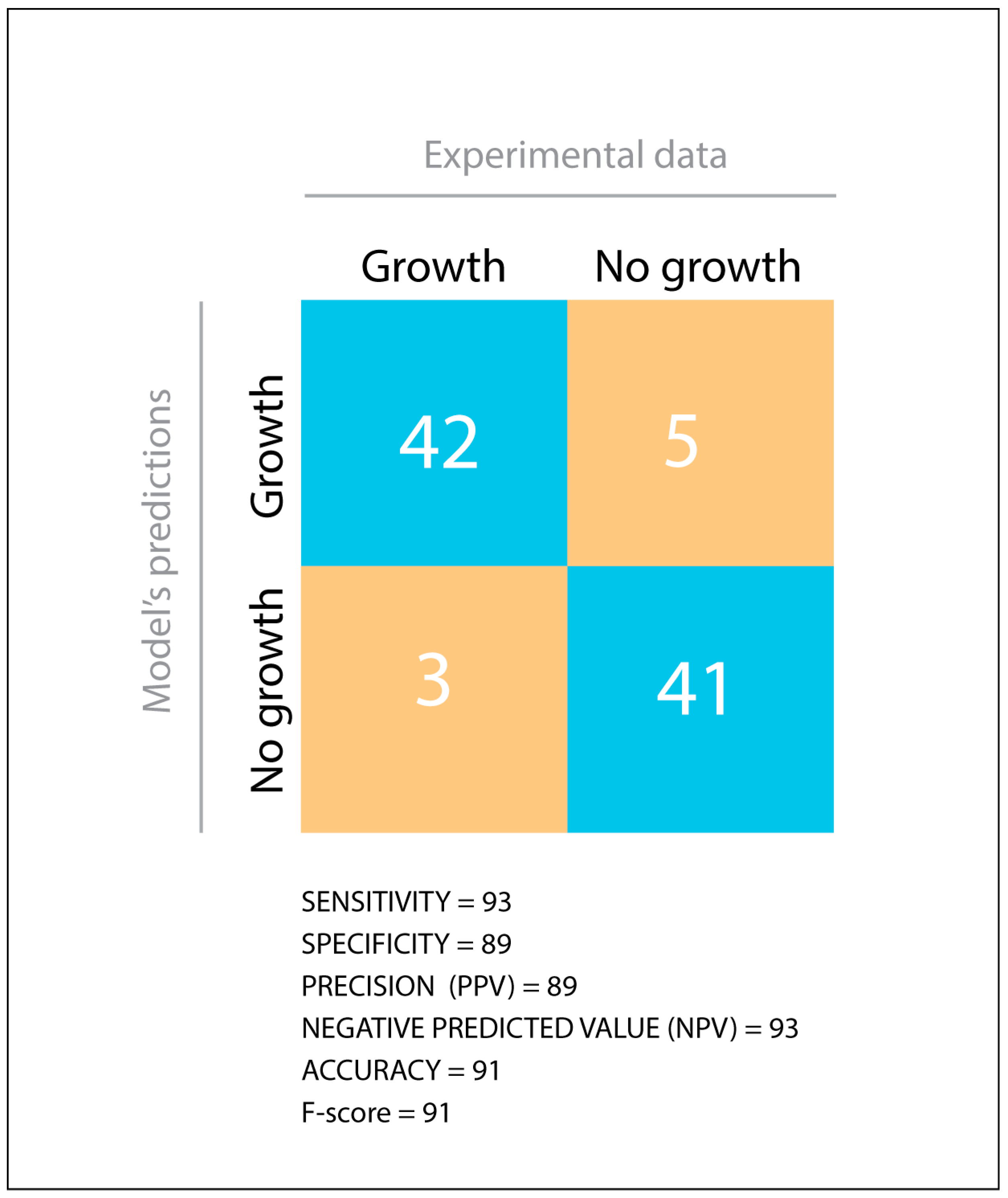
2.9. Model’s Predictive Value Estimation
2.10. Single Gene Deletion Analysis
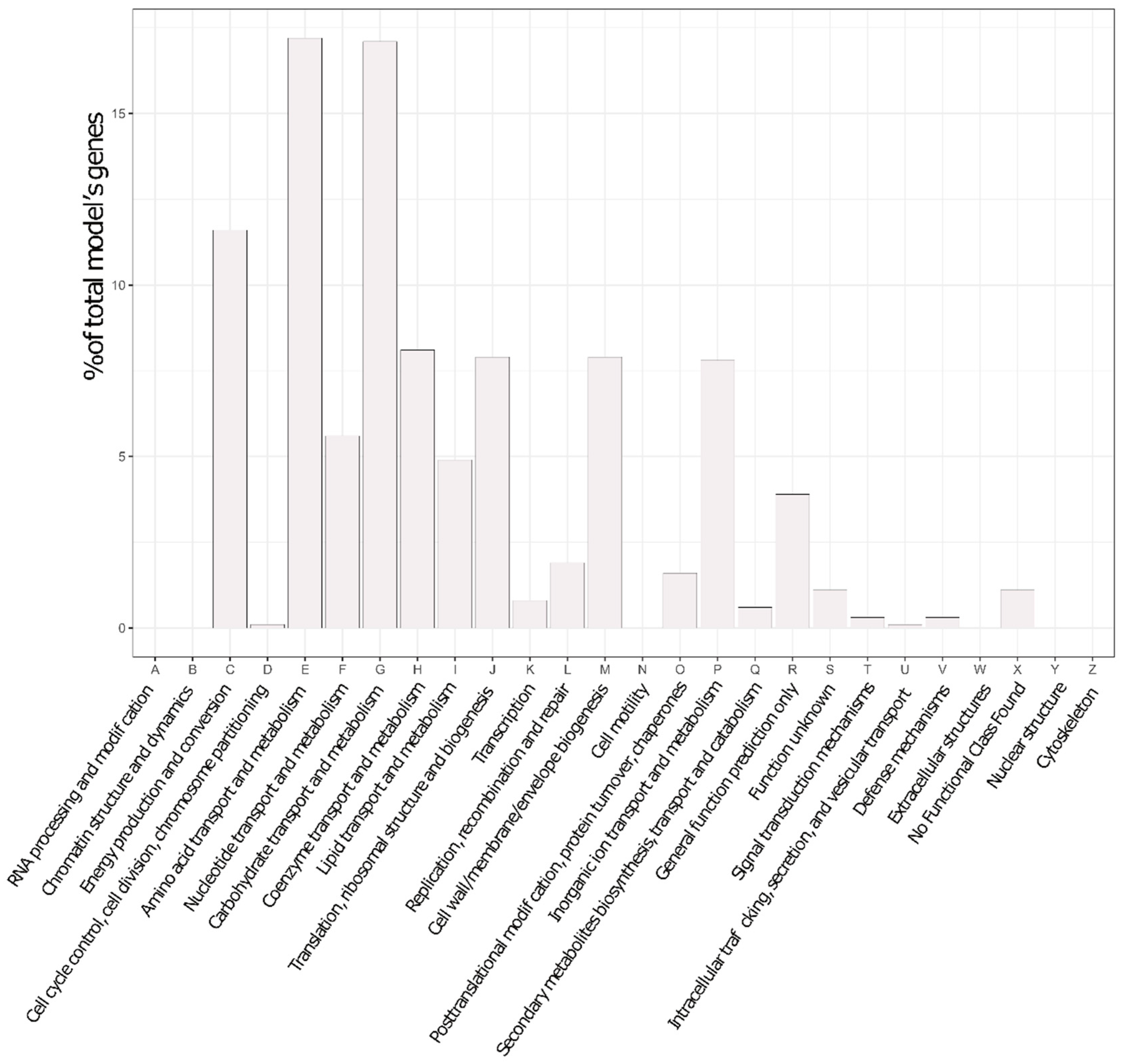
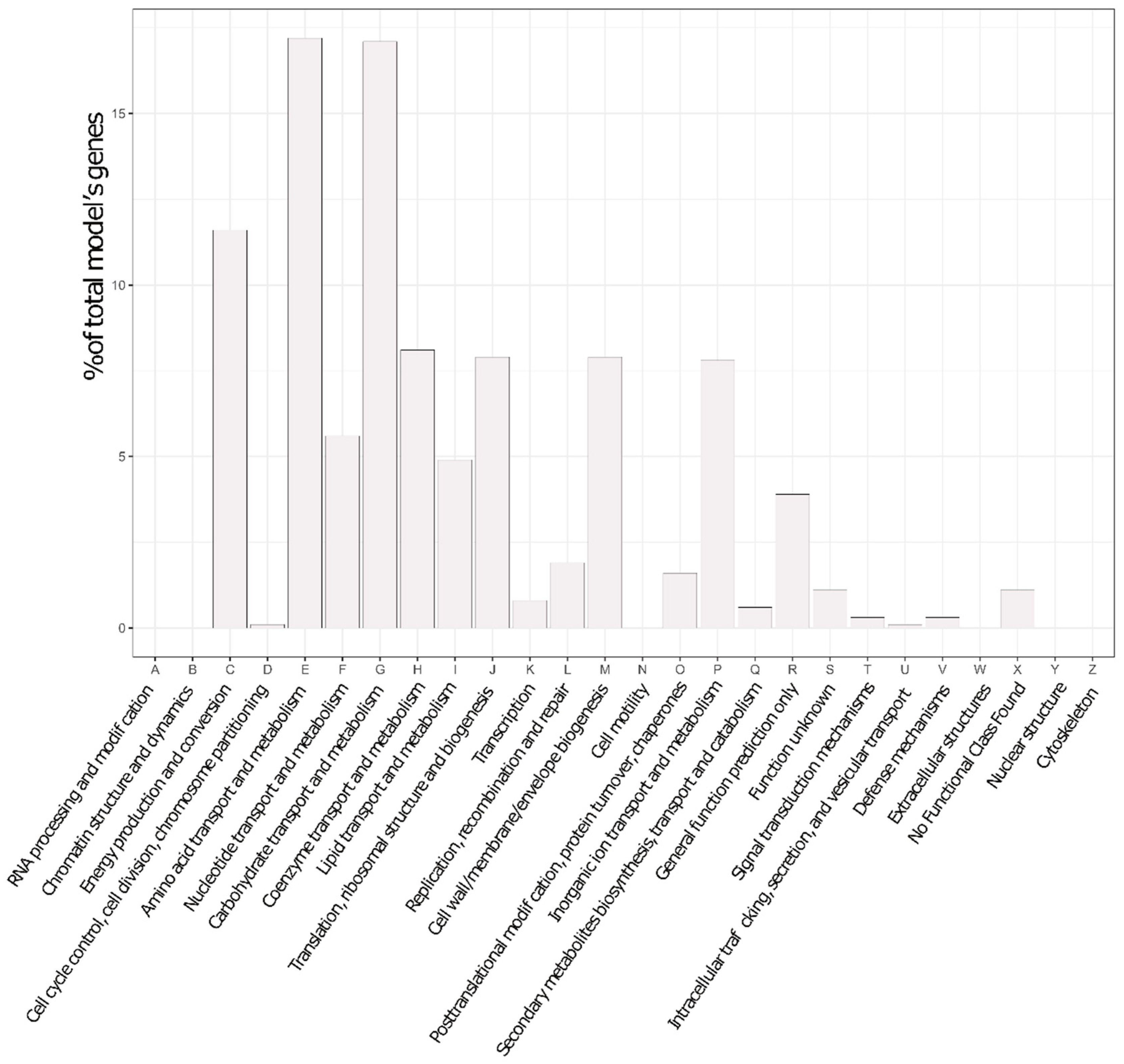
2.11. COG Analyses
3. Results
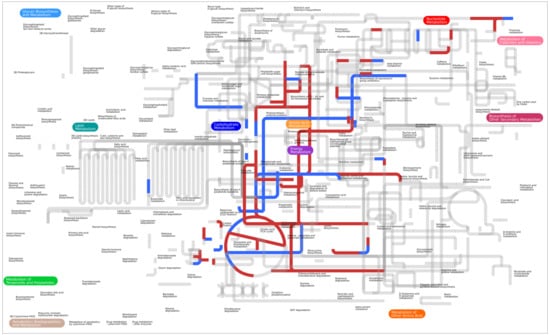
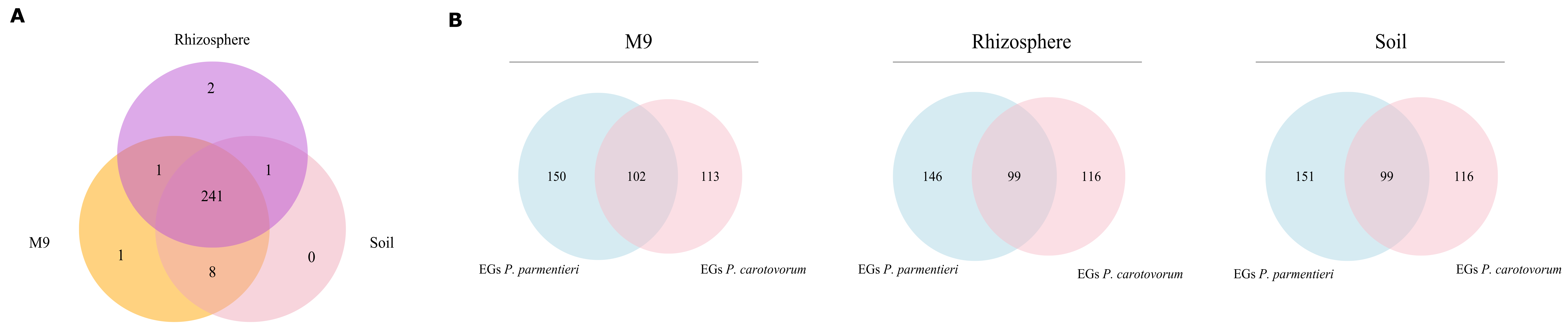
3.1. Validation of a Genome-Scale Metabolic Model of P. parmentieri SCC3193
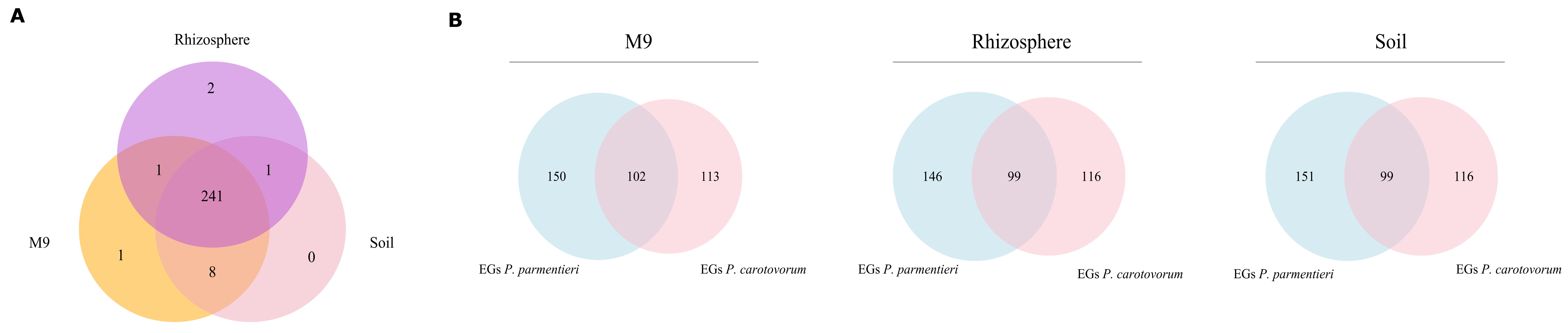
3.2. Phenotypic Characterization of P. parmentieri SCC3193 and Metabolic Model In-Depth Validation
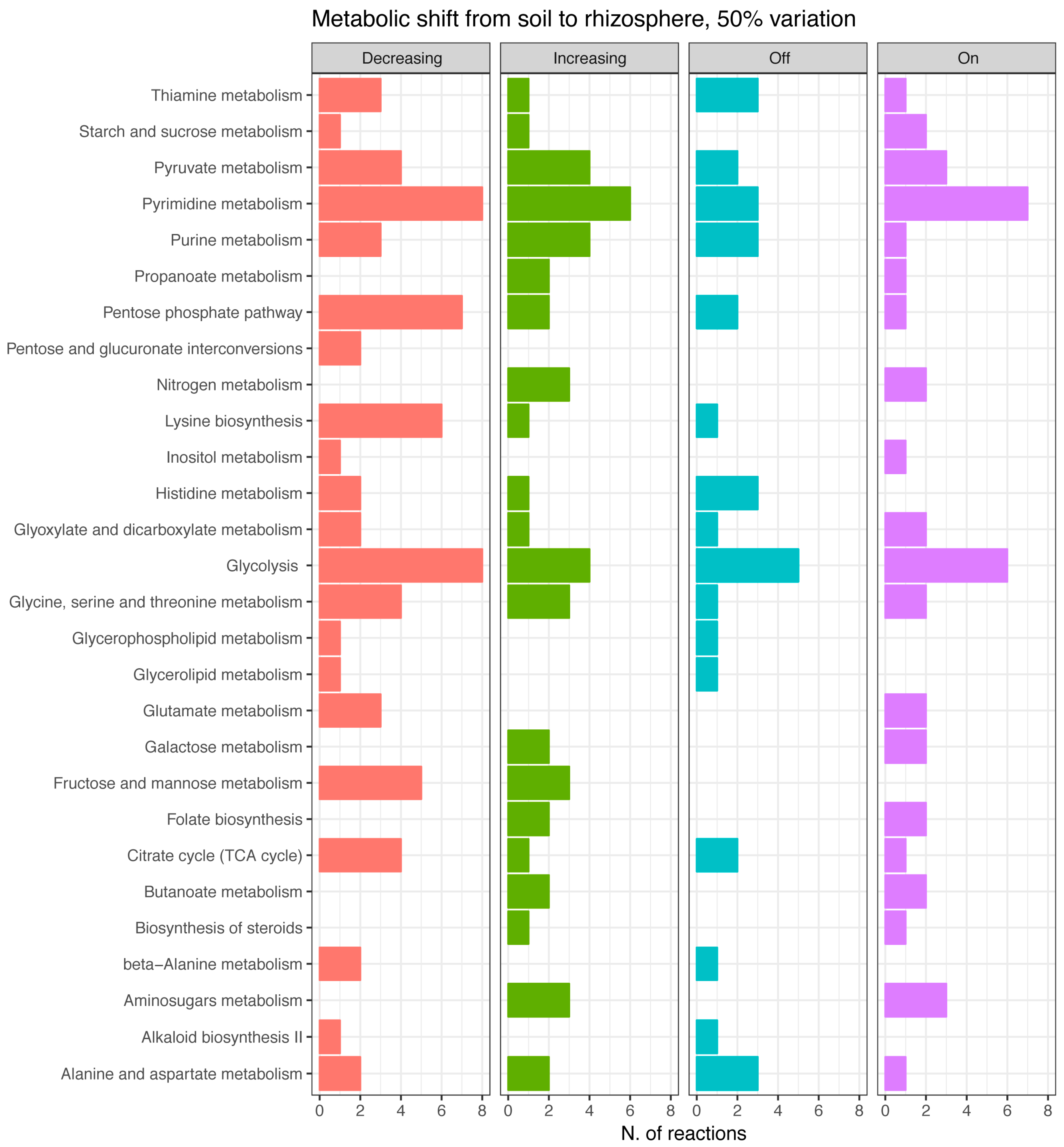
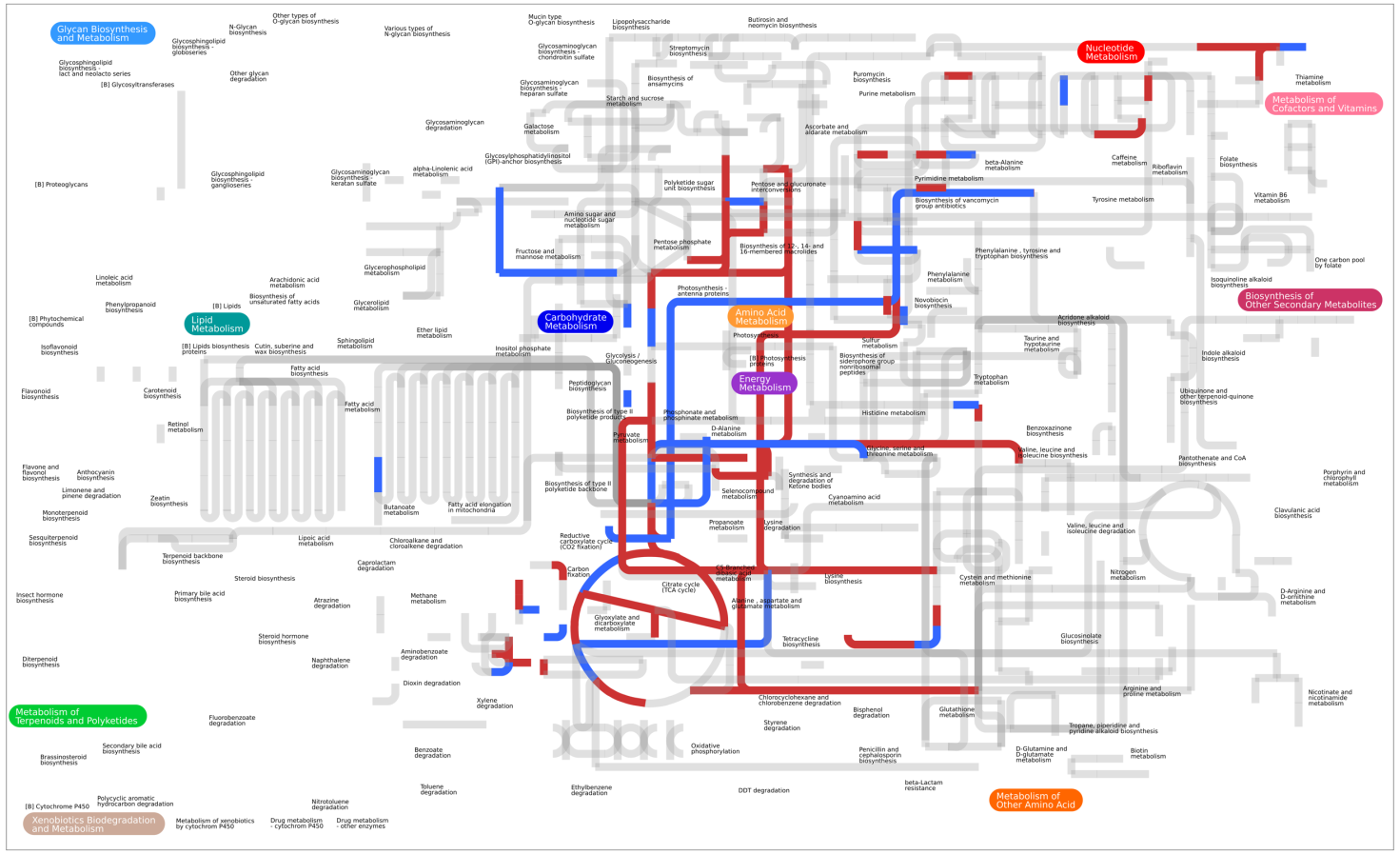
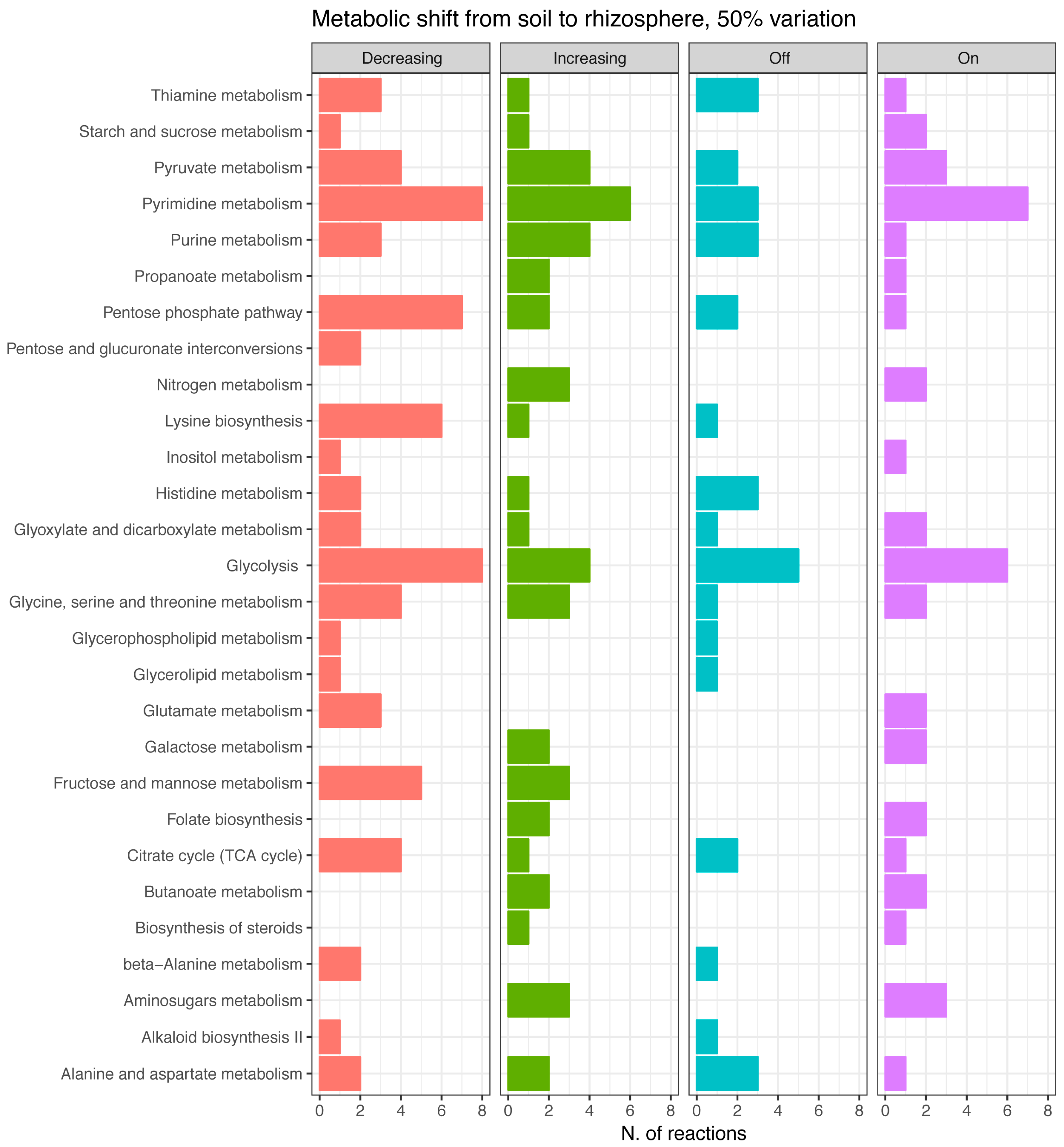
3.3. Differential Metabolic Adaptation to Soil and Rhizosphere Environment
Soil and Rhizosphere Adaptation, a Comparison with the Sinorhizobium meliloti Model
3.4. In Silico Gene Deletions Provide Insight into the Fitness Relevance of Metabolic Modules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Xiang, T.; Zhou, J.-M. Plant immunity: A lesson from pathogenic bacterial effector proteins. Cell. Microbiol. 2009, 11, 1453–1461. [Google Scholar] [CrossRef]
- Mithani, A.; Hein, J.; Preston, G.M. Comparative analysis of metabolic networks provides insight into the evolution of plant pathogenic and nonpathogenic lifestyles in Pseudomonas. Mol. Biol. Evol. 2011, 28, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Rico, A.; Preston, G.M. Pseudomonas syringae pv. tomato DC3000 uses constitutive and apoplast-induced nutrient assimilation pathways to catabolize nutrients that are abundant in the tomato apoplast. Mol. Plant Microbe Interact. 2008, 21, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Motyka, A.; Zoledowska, S.; Sledz, W.; Lojkowska, E. Molecular methods as tools to control plant diseases caused by Dickeya and Pectobacterium spp: A minireview. New Biotechnol. 2017, 39, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, S.T.; Coaker, G.; Day, B.; Staskawicz, B.J. Host-Microbe Interactions: Shaping the Evolution of the Plant Immune Response. Cell 2006, 124, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Põllumaa, L.; Alamäe, T.; Mäe, A. Quorum sensing and expression of virulence in Pectobacteria. Sensors 2012, 12, 3327–3349. [Google Scholar] [CrossRef]
- Adeolu, M.; Seema, A.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morgane. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar]
- Khayi, S.; Cigna, J.; Chong, T.M.; Quêtu-Laurent, A.; Chan, K.-G.; Hélias, V.; Faure, D. Transfer of the potato plant isolates of Pectobacterium wasabiae to Pectobacterium parmentieri sp. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5379–5383. [Google Scholar]
- Toth, I.K.; Bell, K.S.; Holeva, M.C.; Birch, P.R.J. Soft rot Erwiniae: From genes to genomes. Mol. Plant Pathol. 2003, 4, 17–30. [Google Scholar] [CrossRef]
- Pérombelon, M.C.M. Potato diseases caused by soft rot erwinias: An overview of pathogenesis. Plant Pathol. 2002, 51, 1–12. [Google Scholar] [CrossRef]
- Ma, B.; Hibbing, M.E.; Kim, H.-S.; Reedy, R.M.; Yedidia, I.; Breuer, J.J.J.; Breuer, J.J.J.; Glasner, J.D.; Perna, N.T.; Kelman, A.; et al. Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 2007, 97, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Charkowski, A.; Blanco, C.; Condemine, G.; Expert, D.; Franza, T.; Hayes, C.; Hugouvieux-Cotte-Pattat, N.; Solanilla, E.L.; Low, D.; Moleleki, L.; et al. The Role of Secretion Systems and Small Molecules in Soft-Rot Enterobacteriaceae Pathogenicity. Annu. Rev. Phytopathol. 2012, 50, 425–449. [Google Scholar] [CrossRef]
- Zoledowska, S.; Motyka, A.; Zukowska, D.; Sledz, W.; Lojkowska, E. Population Structure and Biodiversity of Pectobacterium parmentieri Isolated from Potato Fields in Temperate Climate. Plant Dis. 2018, 102, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Mansflieds, J.; Genin, S.; Magor, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, R.; Pérombelon, M.C.M.; van Veen, J.A.; van der Wolf, J.M. Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: A review. Plant Pathol. 2011, 60, 999–1013. [Google Scholar] [CrossRef]
- Liu, H.; Coulthurst, S.J.; Pritchard, L.; Hedley, P.E.; Ravensdale, M.; Humphris, S.; Burr, T.; Takle, G.; Brurberg, M.-B.; Birch, P.R.J.; et al. Quorum sensing coordinates brute force and stealth modes of infection in the plant pathogen Pectobacterium atrosepticum. PLoS Pathog. 2008, 4, e1000093. [Google Scholar] [CrossRef]
- Effantin, G.G.; Rivasseau, C.; Gromova, M.; Bligny, R.; Hugouvieux-Cotte-Pattat, N. Massive production of butanediol during plant infection by phytopathogenic bacteria of the genera Dickeya and Pectobacterium. Mol. Microbiol. 2011, 82, 988–997. [Google Scholar] [CrossRef]
- Oberhardt, M.A.; Palsson, B.Ø.; Papin, J.A. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009, 5, 320. [Google Scholar] [CrossRef]
- Duan, G.; Christian, N.; Schwachtje, J.; Walther, D.; Ebenhöh, O. The Metabolic Interplay between Plants and Phytopathogens. Metabolites 2013, 3, 1–23. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Großkopf, T.; Consuegra, J.; Gaffé, J.; Willison, J.C.; Lenski, R.E.; Soyer, O.S.; Schneider, D. Metabolic modelling in a dynamic evolutionary framework predicts adaptive diversification of bacteria in a long-term evolution experiment. BMC Evol. Biol. 2016, 16, 163. [Google Scholar] [CrossRef] [PubMed]
- Angione, C.; Lió, P.; Dobzhansky, T.; Romero, I.G.; Ruvinsky, I.; Gilad, Y.; Fraser, H.B.; Pál, C.; Papp, B.; Lercher, M.J.; et al. Predictive analytics of environmental adaptability in multi-omic network models. Sci. Rep. 2015, 5, 15147. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Bosi, E.; Giudice, A.L.; Fani, R. A Systems Biology View on Bacterial Response to Temperature Shift. In Biotechnology of Extremophiles; Springer: Cham, Switzerland, 2016; pp. 597–618. ISBN 978-3-319-13520-5. [Google Scholar]
- diCenzo, G.C.; Checcucci, A.; Bazzicalupo, M.; Mengoni, A.; Viti, C.; Dziewit, L.; Finan, T.M.; Galardini, M.; Fondi, M.; Harrison, P.W.; et al. Metabolic modelling reveals the specialization of secondary replicons for niche adaptation in Sinorhizobium meliloti. Nat. Commun. 2016, 7, 12219. [Google Scholar] [CrossRef]
- Wang, C.; Deng, Z.-L.; Xie, Z.-M.; Chu, X.-Y.; Chang, J.-W.; Kong, D.-X.; Li, B.-J.; Zhang, H.-Y.; Chen, L.-L. Construction of a genome-scale metabolic network of the plant pathogen Pectobacterium carotovorum provides new strategies for bactericide discovery. FEBS Lett. 2015, 589, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, R.; Cottret, L.; Marmiesse, L.; Gouzy, J.; Genin, S.; Brown, S.; Palmer, K.; Whiteley, M.; Eisenreich, W.; Dandekar, T.; et al. A Resource Allocation Trade-Off between Virulence and Proliferation Drives Metabolic Versatility in the Plant Pathogen Ralstonia solanacearum. PLoS Pathog. 2016, 12, e1005939. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, J.P.; Laine, P.; Niemi, O.; Nykyri, J.; Harjunpää, H.; Auvinen, P.; Paulin, L.; Pirhonen, M.; Palva, T.; Holm, L.; et al. Genome sequence of Pectobacterium sp. strain SCC3193. J. Bacteriol. 2012, 194, 6004. [Google Scholar] [CrossRef]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Nykyri, J.; Niemi, O.; Koskinen, P.; Nokso-Koivisto, J.; Pasanen, M.; Broberg, M.; Plyusnin, I.; Törönen, P.; Holm, L.; Pirhonen, M.; et al. Revised phylogeny and novel horizontally acquired virulence determinants of the model soft rot phytopathogen Pectobacterium wasabiae SCC3193. PLoS Pathog. 2012, 8, e1003013. [Google Scholar] [CrossRef]
- Galardini, M.; Mengoni, A.; Biondi, E.G.; Semeraro, R.; Florio, A.; Bazzicalupo, M.; Benedetti, A.; Mocali, S. DuctApe: A suite for the analysis and correlation of genomic and OmniLogTM Phenotype Microarray data. Genomics 2014, 103, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Segre, D.; Vitkup, D.; Church, G.M.; Segrè, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef]
- Reed, J.L.; Vo, T.D.; Schilling, C.H.; Palsson, B.O. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 2003, 4, R54. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, S.N.; Cañón, P.M.; Contreras, Á.; Ribbeck, M.; Agosín, E. Genome-Scale Reconstruction of the Metabolic Network in Oenococcus oeni to Assess Wine Malolactic Fermentation. Front. Microbiol. 2017, 8, 534. [Google Scholar] [CrossRef] [PubMed]
- Juty, N.; Le Novere, N.; Laibe, C. Identifiers.org and MIRIAM Registry: Community resources to provide persistent identification. Nucleic Acids Res. 2012, 40, D580–D586. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, J.; Jensen, L.E.; Nybroe, O. Soil and rhizosphere as habitats for Pseudomonas inoculants: New knowledge on distribution, activity and physiological state derived from micro-scale and single-cell studies. Plant Soil 2001, 232, 97–108. [Google Scholar] [CrossRef]
- Gorshkov, V.; Kwenda, S.; Petrova, O.; Osipova, E.; Gogolev, Y.; Moleleki, L.N. Global gene expression analysis of cross-protected phenotype of Pectobacterium atrosepticum. PLoS ONE 2017, 12, e0169536. [Google Scholar] [CrossRef]
- Kanchiswamy, C.N.; Malnoy, M.; Maffei, M.E. Chemical diversity of microbial volatiles and their potential for plant growth and productivity. Front. Plant Sci. 2015, 6, 151. [Google Scholar] [CrossRef]
- diCenzo, G.C.; Finan, T.M. Genetic redundancy is prevalent within the 6.7 Mb Sinorhizobium meliloti genome. Mol. Genet. Genom. 2015, 290, 1345–1356. [Google Scholar] [CrossRef]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G.; et al. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018, 46, D1121–D1127. [Google Scholar] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.J.; Cai, J.; Wang, L.; Simons-Senftle, M.N.; Maranas, C.D. Standardizing biomass reactions and ensuring complete mass balance in genome scale metabolic models. Bioinformatics 2017, 33, 3603–3609. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P. parmentieri SCC3193 Genome | |
|---|---|
| Total genome size | 4821 |
| Total protein-coding genes | 4449 |
| Pseudo Genes | 266 |
| iLP1245 characteristics | |
| Total genes of P. parmentieri SCC3193 | 1245 |
| Genes | 1224 |
| Pseudo Genes | 21 |
| Gap-filling genes | 7 |
| Total reactions | 2182 |
| Gene-associated reactions | 2120 |
| Exchange reactions | 195 |
| Transport reactions | 324 |
| Spontaneous reactions | 55 |
| Total metabolites | 2080 |
| Locus Gene | Candidate Drug Targets | Function | EC No. |
|---|---|---|---|
| W5S_RS00975 | WP_012822018.1 | 5,10-methylenetetrahydrofolate reductase | 1.5.1.20 |
| W5S_RS01035 | WP_012822025.1 | UDP-N-acetylenolpyruvoylglucosamine reductase | 1.1.1.158 |
| W5S_RS01045 | WP_012822027.1 | pantothenate kinase | 2.7.1.33 |
| W5S_RS09185 | WP_014699609.1 | GTP cyclohydrolase I | 3.5.4.16 |
| W5S_RS10395 | WP_014699824.1 | NAD synthetase | 6.3.1.5 |
| W5S_RS13505 | WP_014700413.1 | Thymidylate kinase | 2.7.4.9 |
| W5S_RS06665 | WP_014699128.1 | bifunctional folylpolyglutamate synthase/ dihydrofolate synthase | 6.3.2.12/6.3.2.17 |
| W5S_RS18995 | WP_014701427.1 | UDP-diphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase | 2.4.1.227 |
| W5S_RS19920 | WP_014701595.1 | methionine synthase | 2.1.2.13 |
| W5S_RS20855 | WP_005969274.1 | aspartate-semialdehyde dehydrogenase | 1.2.1.11 |
| W5S_RS22025 | WP_014701962.1 | phosphopantetheine adenylyltransferase | 2.7.7.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoledowska, S.; Presta, L.; Fondi, M.; Decorosi, F.; Giovannetti, L.; Mengoni, A.; Lojkowska, E. Metabolic Modeling of Pectobacterium parmentieri SCC3193 Provides Insights into Metabolic Pathways of Plant Pathogenic Bacteria. Microorganisms 2019, 7, 101. https://doi.org/10.3390/microorganisms7040101
Zoledowska S, Presta L, Fondi M, Decorosi F, Giovannetti L, Mengoni A, Lojkowska E. Metabolic Modeling of Pectobacterium parmentieri SCC3193 Provides Insights into Metabolic Pathways of Plant Pathogenic Bacteria. Microorganisms. 2019; 7(4):101. https://doi.org/10.3390/microorganisms7040101
Chicago/Turabian StyleZoledowska, Sabina, Luana Presta, Marco Fondi, Francesca Decorosi, Luciana Giovannetti, Alessio Mengoni, and Ewa Lojkowska. 2019. "Metabolic Modeling of Pectobacterium parmentieri SCC3193 Provides Insights into Metabolic Pathways of Plant Pathogenic Bacteria" Microorganisms 7, no. 4: 101. https://doi.org/10.3390/microorganisms7040101
APA StyleZoledowska, S., Presta, L., Fondi, M., Decorosi, F., Giovannetti, L., Mengoni, A., & Lojkowska, E. (2019). Metabolic Modeling of Pectobacterium parmentieri SCC3193 Provides Insights into Metabolic Pathways of Plant Pathogenic Bacteria. Microorganisms, 7(4), 101. https://doi.org/10.3390/microorganisms7040101