Insights into the Bacterial Profiles and Resistome Structures Following the Severe 2018 Flood in Kerala, South India


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
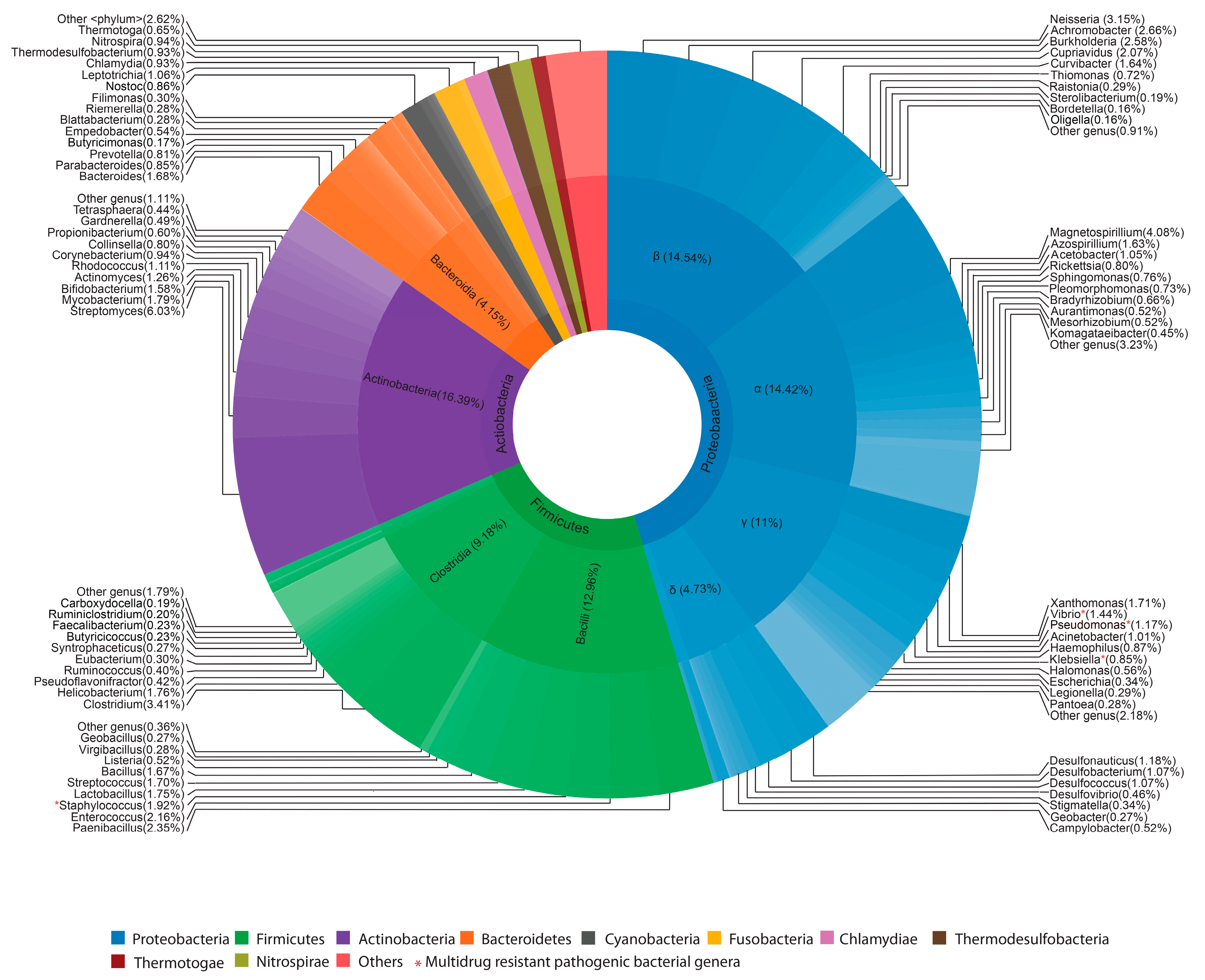
2.1. Bacterial Profiles of Extremely Flooded Sites
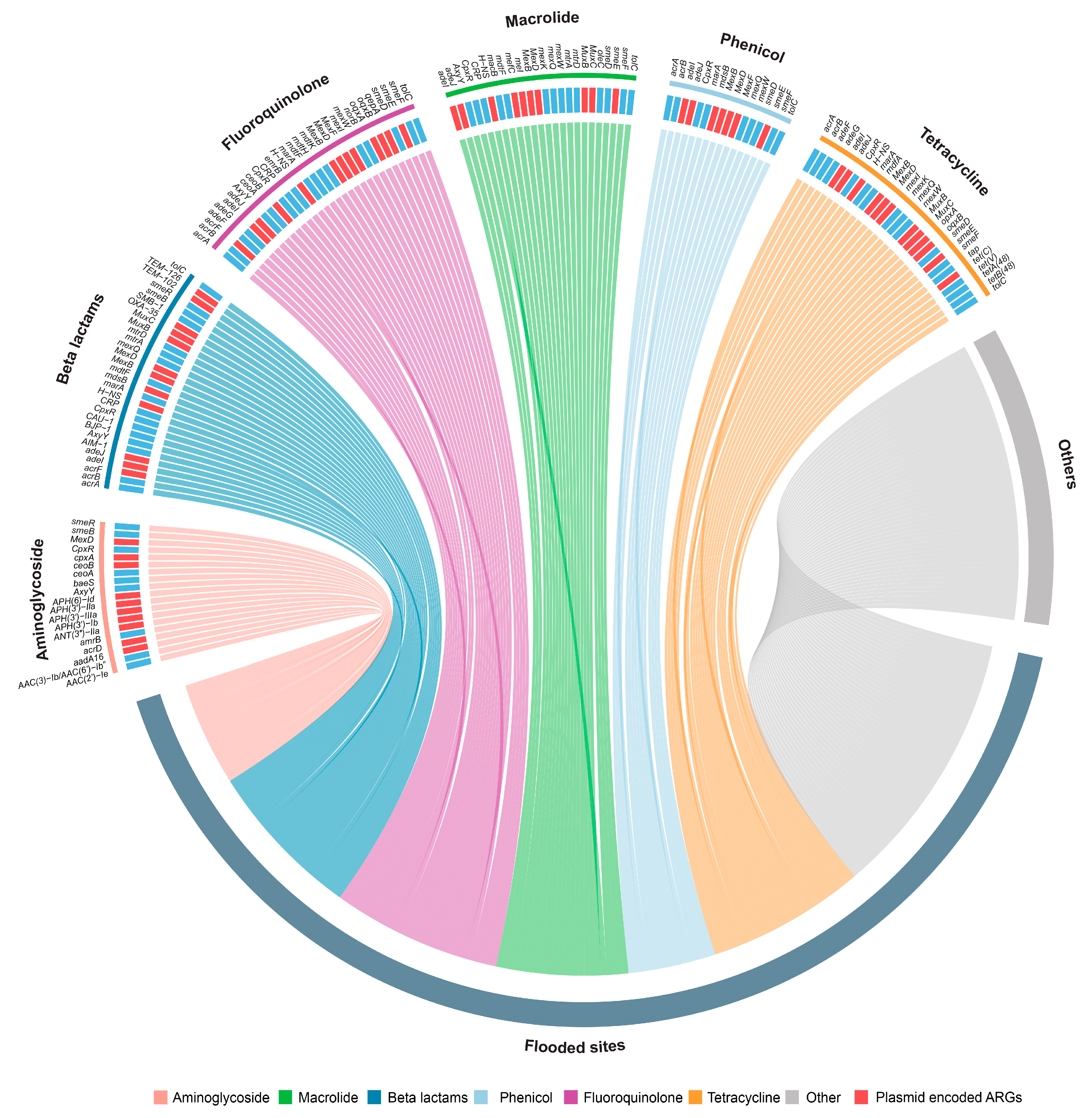
2.2. Resistome of Flooded Sites
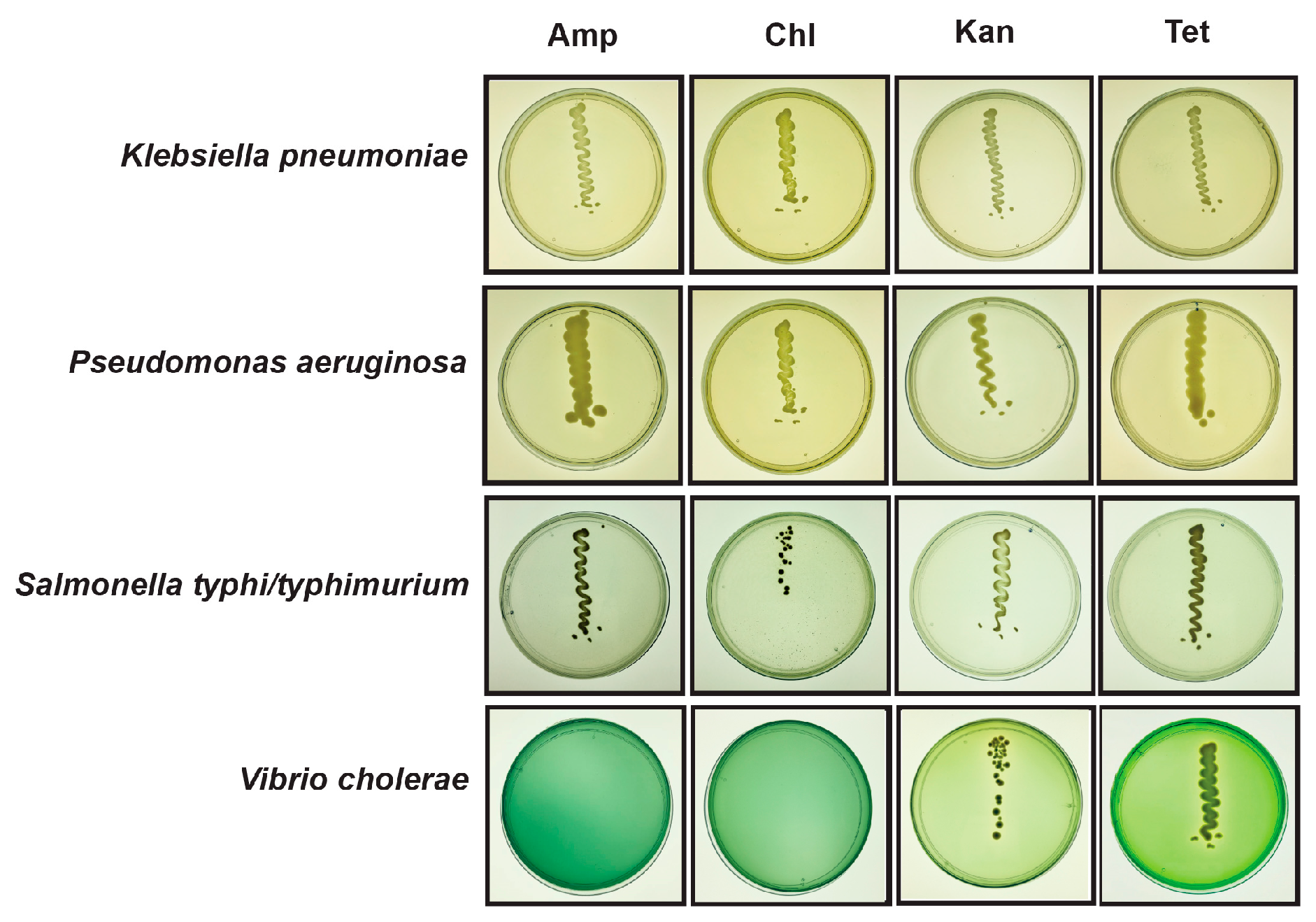
2.3. Viability of Pathogenic and Resistant Bacteria in Flooded Sites
3. Materials and Methods
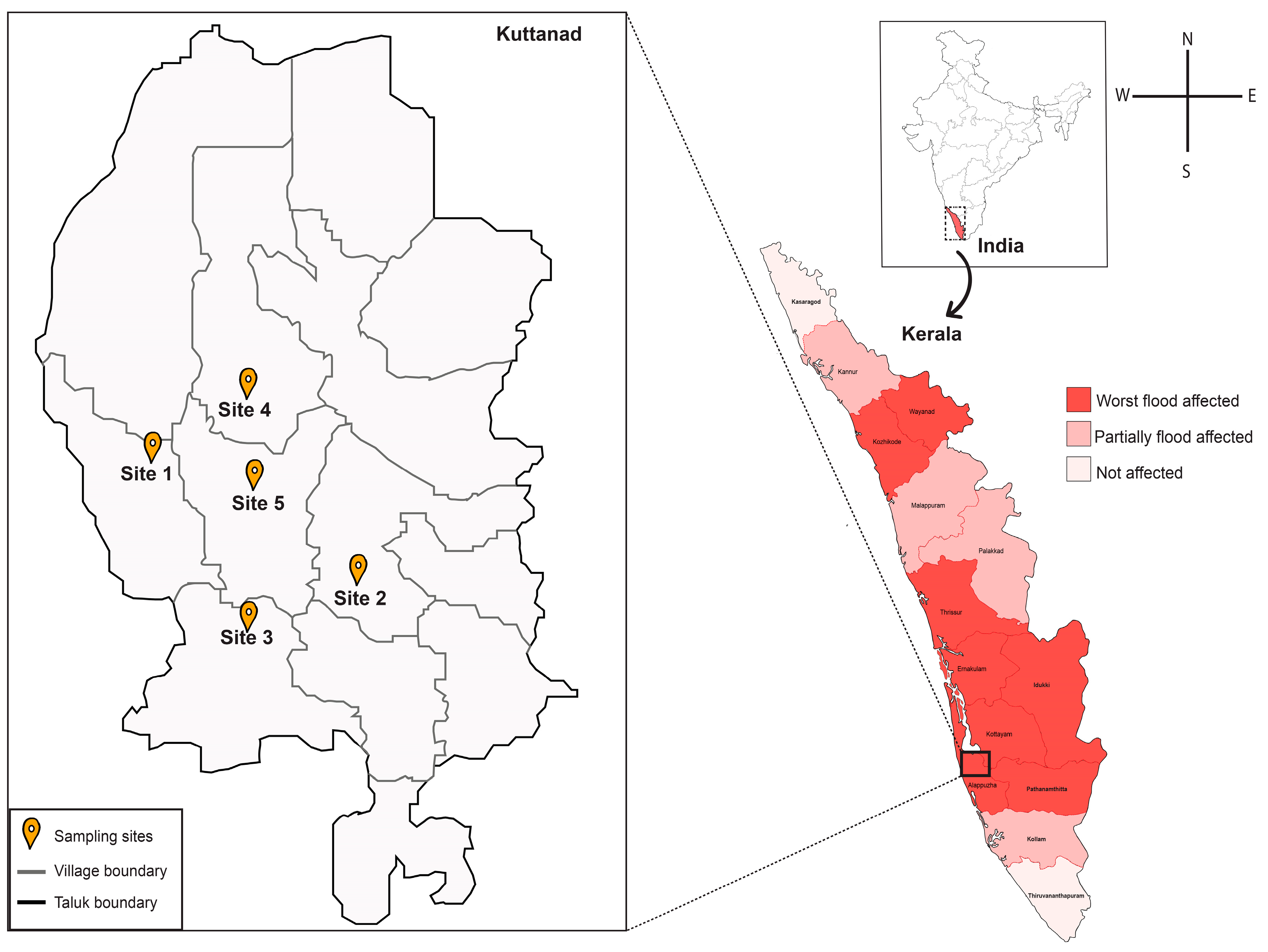
3.1. Sample Collection
3.2. DNA Extraction
3.3. Shotgun Metagenomic Sequencing and Analysis
3.4. Abundance of Selected Pathogens
3.5. Identification of Antibiotic-Resistant Bacterial Pathogens
3.6. 16S rRNA Sequencing
3.7. Availability of Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Z.-D.; Li, J.; Zhang, Y.; Ding, G.-Y.; Xu, X.; Gao, L.; Liu, X.-N.; Liu, Q.-Y.; Jiang, B.-F. Distributed lag effects and vulnerable groups of floods on bacillary dysentery in Huaihua, China. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kerala Floods 2018 Joint Detailed Needs Assessment Report. Available online: https://reliefweb.int/report/india/kerala-floods-2018-joint-detailed-needs-assessment-report (accessed on 13 July 2019).
- Nowfal, N.; Sarath, S. Effective Disaster Management Mechanism: Experience from Kerala Floods. Univers. Rev. 2018, 7, 503–508. [Google Scholar]
- Agarwal, R. Lesson Learned from Killer Floods in Kerala: Time for Retrospection. Manag. Econ. Res. J. 2018, 4, 268–280. [Google Scholar] [CrossRef]
- Study Report, Kerala Floods of August 2018 (September, 2018). Available online: https://reliefweb.int/report/india/study-report-kerala-floods-august-2018-september-2018 (accessed on 13 July 2019).
- Alderman, K.; Turner, L.R.; Tong, S. Floods and human health: A systematic review. Env. Int. 2012, 47, 37–47. [Google Scholar] [CrossRef]
- Pandey, P.K.; Kass, P.H.; Soupir, M.L.; Biswas, S.; Singh, V.P. Contamination of water resources by pathogenic bacteria. Amb. Express. 2014, 4, 51:1–51:16. [Google Scholar] [CrossRef]
- Taylor, J.; man Lai, K.; Davies, M.; Clifton, D.; Ridley, I.; Biddulph, P. Flood management: Prediction of microbial contamination in large-scale floods in urban environments. Env. Int. 2011, 37, 1019–1029. [Google Scholar] [CrossRef]
- Laffite, A.; Kilunga, P.I.; Kayembe, J.M.; Devarajan, N.; Mulaji, C.K.; Giuliani, G.; Slaveykova, V.I.; Poté, J. Hospital effluents are one of several sources of metal, antibiotic resistance genes, and bacterial markers disseminated in Sub-Saharan urban rivers. Front Microbiol. 2016, 7, 1128:1–1128:14. [Google Scholar] [CrossRef]
- Rodgers, K.; McLellan, I.; Peshkur, T.; Williams, R.; Tonner, R.; Hursthouse, A.S.; Knapp, C.W.; Henriquez, F.L. Can the legacy of industrial pollution influence antimicrobial resistance in estuarine sediments. Env. Chem. Lett. 2019, 17, 595–607. [Google Scholar] [CrossRef]
- Czekalski, N.; Berthold, T.; Caucci, S.; Egli, A.; Bürgmann, H. Increased levels of multiresistant bacteria and resistance genes after wastewater treatment and their dissemination into Lake Geneva, Switzerland. Front Microbiol. 2012, 3, 106:1–106:18. [Google Scholar] [CrossRef]
- Pruden, A.; Arabi, M.; Storteboom, H.N. Correlation between upstream human activities and riverine antibiotic resistance genes. Env. Sci. Technol. 2012, 46, 11541–11549. [Google Scholar] [CrossRef]
- Berkner, S.; Konradi, S.; Schönfeld, J. Antibiotic resistance and the environment—there and back again. Embo. Rep. 2014, 15, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Sanganyado, E.; Gwenzi, W. Antibiotic resistance in drinking water systems: Occurrence, removal, and human health risks. Sci. Total Env. 2019, 669, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.J. Environmental factors influencing the development and spread of antibiotic resistance. Fems. Microbiol. Rev. 2017, 42, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.-R. Megacities as sources for pathogenic bacteria in rivers and their fate downstream. Int. J. Microbiol. 2010, 2011, 798292:1–798292:13. [Google Scholar] [CrossRef]
- Stalder, T.; Barraud, O.; Casellas, M.; Dagot, C.; Ploy, M.-C. Integron involvement in environmental spread of antibiotic resistance. Front Microbiol. 2012, 3, 119:1–119:14. [Google Scholar] [CrossRef]
- Amaral-Zettler, L.A.; Rocca, J.D.; Lamontagne, M.G.; Dennett, M.R.; Gast, R.J. Changes in microbial community structure in the wake of Hurricanes Katrina and Rita. Env. Sci. Technol. 2008, 42, 9072–9078. [Google Scholar] [CrossRef]
- Gowrisankar, G.; Chelliah, R.; Ramakrishnan, S.R.; Elumalai, V.; Dhanamadhavan, S.; Brindha, K.; Antony, U.; Elango, L. Chemical, microbial and antibiotic susceptibility analyses of groundwater after a major flood event in Chennai. Sci. Data. 2017, 4, 170135:1–170135:13. [Google Scholar] [CrossRef]
- Yu, P.; Zaleski, A.; Li, Q.; He, Y.; Mapili, K.; Pruden, A.; Alvarez, P.J.; Stadler, L.B. Elevated Levels of Pathogenic Indicator Bacteria and Antibiotic Resistance Genes after Hurricane Harvey’s Flooding in Houston. Env. Sci. Technol Lett. 2018, 5, 481–486. [Google Scholar] [CrossRef]
- Mhuantong, W.; Wongwilaiwalin, S.; Laothanachareon, T.; Eurwilaichitr, L.; Tangphatsornruang, S.; Boonchayaanant, B.; Limpiyakorn, T.; Pattaragulwanit, K.; Punmatharith, T.; McEvoy, J. Survey of microbial diversity in flood areas during Thailand 2011 flood crisis using high-throughput tagged amplicon pyrosequencing. PLoS ONE 2015, 10, e0128043:1–e0128043:20. [Google Scholar] [CrossRef]
- Milly, P.C.D.; Wetherald, R.T.; Dunne, K.; Delworth, T.L. Increasing risk of great floods in a changing climate. Nature 2002, 415, 514–517. [Google Scholar] [CrossRef]
- Reilly, T.J.; Focazio, M.J.; Simmons, D.L. Resetting the bar: Establishing baselines for persistent contaminants after Hurricane Sandy in the coastal environments of New Jersey and New York, USA. Mar. Pollut Bull. 2016, 107, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Kabeer, R.; Varghese, R.; Sylas, V. Rhizosphere of water hyacinth as a niche for multidrug resistant Aeromonas taiwanensis and Paenibacillus taiwanensis: A study from a tropical wetland of South India. Rhizosphere 2018, 6, 20–22. [Google Scholar] [CrossRef]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Adv. Infect Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hassard, F.; Gwyther, C.L.; Farkas, K.; Andrews, A.; Jones, V.; Cox, B.; Brett, H.; Jones, D.L.; McDonald, J.E.; Malham, S.K. Abundance and distribution of enteric bacteria and viruses in coastal and estuarine sediments—a review. Front Microbiol. 2016, 7, 1692:1–1692:31. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Lucas, L.R.; Montor-Antonio, J.J.; Cortés-López, N.G.; del Moral, S. Strategies for the extraction, purification and amplification of metagenomic DNA from soil growing sugarcane. Adv. Biol. Chem. 2014, 4, 281–289. [Google Scholar] [CrossRef]
- Maniatis, T.; Fritsch, E.; Sambrook, J. Molecular Cloning. A Laboratory Manual; Cold Spring Harbor Laboratory: New York, NY, USA, 1982; ISBN 0-87969-136-0. [Google Scholar]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 19 October 2019).
- Gordon, A.; Hannon, G. Fastx-Toolkit. FASTQ/A Short-Reads Preprocessing Tools (Unpublished). 2010, 5. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 19 October 2019).
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Peer J. Prepr. 2018, 37, 852–857. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, Y.; Yu, B.; Yang, M. Changes of resistome, mobilome and potential hosts of antibiotic resistance genes during the transformation of anaerobic digestion from mesophilic to thermophilic. Water Res. 2016, 98, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Kristiansson, E.; Fick, J.; Janzon, A.; Grabic, R.; Rutgersson, C.; Weijdegård, B.; Söderström, H.; Larsson, D.J. Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PloS ONE 2011, 6, e17038:1–e17038:7. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Imchen, M.; Kumavath, R.; Barh, D.; Azevedo, V.; Ghosh, P.; Viana, M.; Wattam, A.R. Searching for signatures across microbial communities: Metagenomic analysis of soil samples from mangrove and other ecosystems. Sci. Rep. 2017, 7, 8859:1–8859:13. [Google Scholar] [CrossRef]
- Waksman, S.A. Principles of Soil Microbiology; The Williams and Willkins Co.: Baltimore, MD, USA, 1927. [Google Scholar]
- Johnson, L.F.; Curl, E.A.; Bond, J.H.; Fribourg, H.A. Methods for studying soil microflora: Plant disease relationships; Burgess Publishing Co.: Minneapolis, MN, USA, 1960; pp. 5–7. [Google Scholar]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.; Carmeli, Y.; Falagas, M.; Giske, C.; Harbarth, S.; Hindler, J.; Kahlmeter, G.; Olsson--Liljequist, B. Multidrug--resistant, extensively drug--resistant and pandrug--resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Sato, T.; Weightman, A.J.; Martin, T.A.; Fry, J.C.; Hiom, S.J.; Wade, W.G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Env. Microbiol. 1998, 64, 795–799. [Google Scholar]
- Lee, P.Y.; Costumbrado, J.; Hsu, C.-Y.; Kim, Y.H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. 2012, 62, e3923:1–e3923:5. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Okaka, F.O.; Odhiambo, B. Relationship between Flooding and Out Break of Infectious Diseases in Kenya: A Review of the Literature. J. Env. Public Health. 2018, 2018, 5452938:1–5452938:8. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Worldwide prevalence of anaemia 1993–2005. In WHO Global Database on Anaemia; De Benoist, B., Cogswell, M., Egli, I., McLean, E., Eds.; WHO Press: Geneva, Switzerland, 2008; ISBN 978-9-2-415-9665-7. [Google Scholar]
- Garner, E.; Wallace, J.S.; Argoty, G.A.; Wilkinson, C.; Fahrenfeld, N.; Heath, L.S.; Zhang, L.; Arabi, M.; Aga, D.S.; Pruden, A. Metagenomic profiling of historic Colorado Front Range flood impact on distribution of riverine antibiotic resistance genes. Sci. Rep. 2016, 6, 38432. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.; Dantas, G. The shared antibiotic resistome of soil bacteria and human pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Von Wintersdorff, C.J.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.; Wolffs, P.F. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front Microbiol. 2016, 7, 173:1–173:10. [Google Scholar] [CrossRef]
- Lovell-Read, S.R.; de Carvalho, L.P.S. Salmonella, meet mycobacteria. J. Exp. Med. 2019, 216, 721–722. [Google Scholar] [CrossRef]
- Wojcik, O.; Holt, J.; Kjerulf, A.; Müller, L.; Ethelberg, S.; Mølbak, K. Personal protective equipment, hygiene behaviours and occupational risk of illness after July 2011 flood in Copenhagen, Denmark. Epidemiol. Infect. 2013, 141, 1756–1763. [Google Scholar] [CrossRef]
- Yee, E.L.; Palacio, H.; Atmar, R.L.; Shah, U.; Kilborn, C.; Faul, M.; Gavagan, T.E.; Feigin, R.D.; Versalovic, J.; Neill, F.H. Widespread outbreak of norovirus gastroenteritis among evacuees of Hurricane Katrina residing in a large “megashelter” in Houston, Texas: Lessons learned for prevention. Clin. Infect Dis. 2007, 44, 1032–1039. [Google Scholar] [CrossRef]
- WHO Publishes List of Bacteria for which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 30 August 2019).
- Emerson, J.B.; Keady, P.B.; Brewer, T.E.; Clements, N.; Morgan, E.E.; Awerbuch, J.; Miller, S.L.; Fierer, N. Impacts of flood damage on airborne bacteria and fungi in homes after the 2013 Colorado Front Range flood. Env. Sci. Technol. 2015, 49, 2675–2684. [Google Scholar] [CrossRef]
- Narendrakumar, L.; Gupta, S.S.; Johnson, J.B.; Ramamurthy, T.; Thomas, S. Molecular Adaptations and Antibiotic Resistance in Vibrio cholerae: A Communal Challenge. Microb. Drug Resist. 2019, 25, 1012–1022. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaya Divakaran, S.; Sara Philip, J.; Chereddy, P.; Nori, S.R.C.; Jaya Ganesh, A.; John, J.; Nelson-Sathi, S. Insights into the Bacterial Profiles and Resistome Structures Following the Severe 2018 Flood in Kerala, South India. Microorganisms 2019, 7, 474. https://doi.org/10.3390/microorganisms7100474
Jaya Divakaran S, Sara Philip J, Chereddy P, Nori SRC, Jaya Ganesh A, John J, Nelson-Sathi S. Insights into the Bacterial Profiles and Resistome Structures Following the Severe 2018 Flood in Kerala, South India. Microorganisms. 2019; 7(10):474. https://doi.org/10.3390/microorganisms7100474
Chicago/Turabian StyleJaya Divakaran, Soumya, Jamiema Sara Philip, Padma Chereddy, Sai Ravi Chandra Nori, Akshay Jaya Ganesh, Jiffy John, and Shijulal Nelson-Sathi. 2019. "Insights into the Bacterial Profiles and Resistome Structures Following the Severe 2018 Flood in Kerala, South India" Microorganisms 7, no. 10: 474. https://doi.org/10.3390/microorganisms7100474
APA StyleJaya Divakaran, S., Sara Philip, J., Chereddy, P., Nori, S. R. C., Jaya Ganesh, A., John, J., & Nelson-Sathi, S. (2019). Insights into the Bacterial Profiles and Resistome Structures Following the Severe 2018 Flood in Kerala, South India. Microorganisms, 7(10), 474. https://doi.org/10.3390/microorganisms7100474