Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome


{kind=link}
Abstract
1. Introduction
2. Pathogenetic Mechanisms of STEC-HUS
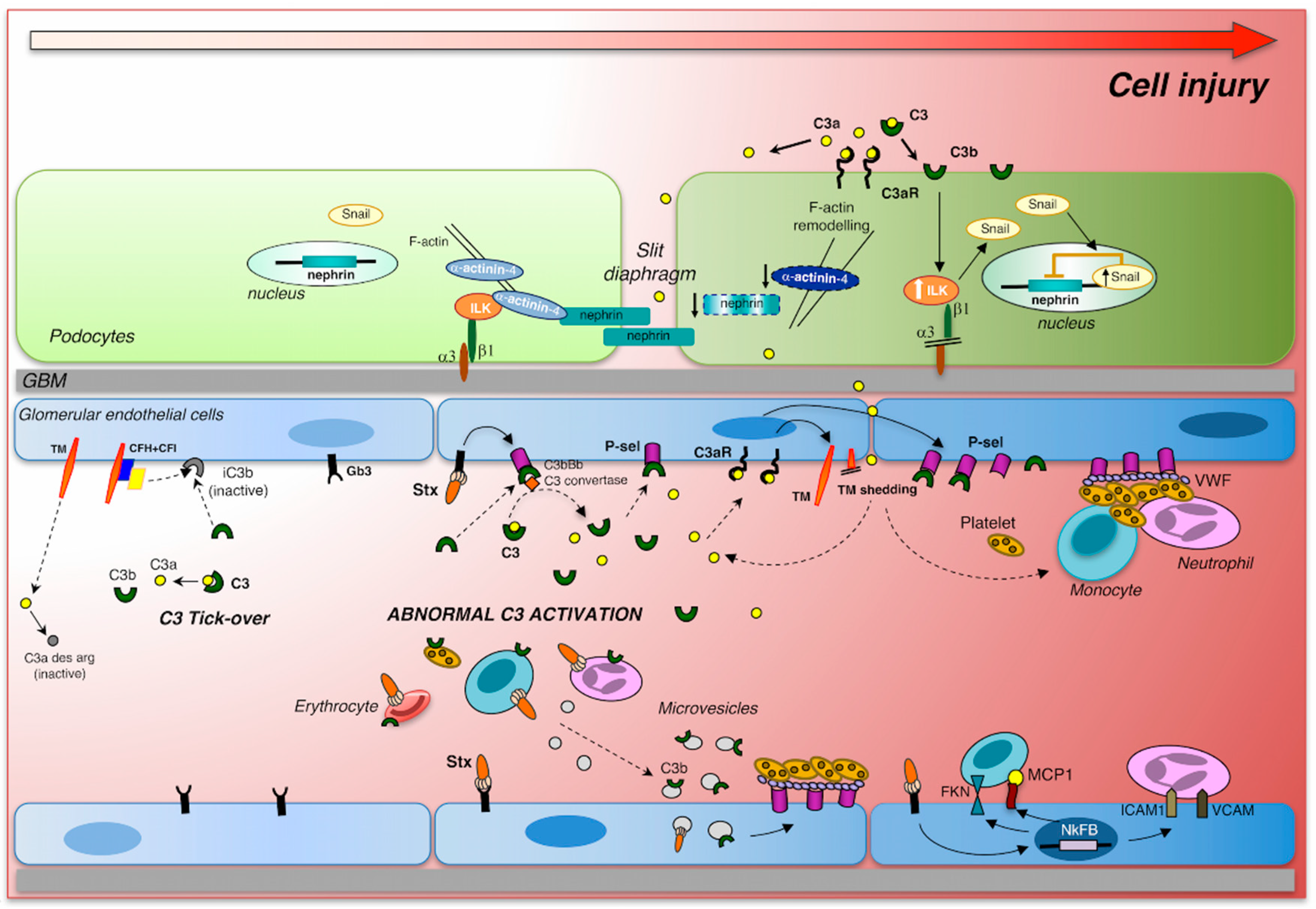
3. Cytotoxic Effect of Shiga Toxin on Glomerular Endothelial Cells and Podocytes
4. The Complement System
5. Stx-associated HUS and Complement Activation: Clinical and Experimental Evidence
6. Complement Activation Induces Glomerular Endothelial Damage
7. Complement Activation Induces Glomerular Podocyte Injury
8. Treatments
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet Lond. Engl. 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005, 16, 1035–1050. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A.; Gannon, V.; Sargeant, J.M. Verocytotoxin-producing Escherichia coli (VTEC). Vet. Microbiol. 2010, 140, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Noris, M.; Remuzzi, G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001, 60, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Majowicz, S.E.; Scallan, E.; Jones-Bitton, A.; Sargeant, J.M.; Stapleton, J.; Angulo, F.J.; Yeung, D.H.; Kirk, M.D. Global incidence of human Shiga toxin-producing Escherichia coli infections and deaths: A systematic review and knowledge synthesis. Foodborne Pathog. Dis. 2014, 11, 447–455. [Google Scholar] [CrossRef]
- Rivas, M.; Chinen, I.; Miliwebsky, E.; Masana, M. Risk Factors for Shiga Toxin-Producing Escherichia coli-Associated Human Diseases. Microbiol. Spectr. 2014, 2, 1–14. [Google Scholar] [CrossRef]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic uraemic syndrome. Lancet Lond. Engl. 2017, 390, 681–696. [Google Scholar] [CrossRef]
- Torres, A.G.; Amaral, M.M.; Bentancor, L.; Galli, L.; Goldstein, J.; Krüger, A.; Rojas-Lopez, M. Recent Advances in Shiga Toxin-Producing Escherichia coli Research in Latin America. Microorganisms 2018, 6, 100. [Google Scholar] [CrossRef]
- Buchholz, U.; Bernard, H.; Werber, D.; Böhmer, M.M.; Remschmidt, C.; Wilking, H.; Deleré, Y.; an der Heiden, M.; Adlhoch, C.; Dreesman, J.; et al. German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 2011, 365, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Köck, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Remuzzi, G. A German outbreak of haemolytic uraemic syndrome. Lancet 2011, 378, 1057–1058. [Google Scholar] [CrossRef]
- Hauswaldt, S.; Nitschke, M.; Sayk, F.; Solbach, W.; Knobloch, J.K.-M. Lessons Learned from Outbreaks of Shiga Toxin Producing Escherichia coli. Curr. Infect. Dis. Rep. 2013, 15, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Kampmeier, S.; Berger, M.; Mellmann, A.; Karch, H.; Berger, P. The 2011 German Enterohemorrhagic Escherichia coli O104:H4 Outbreak-The Danger Is Still Out There. Curr. Top. Microbiol. Immunol. 2018, 416, 117–148. [Google Scholar] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Crowe, S.J.; Bottichio, L.; Shade, L.N.; Whitney, B.M.; Corral, N.; Melius, B.; Arends, K.D.; Donovan, D.; Stone, J.; Allen, K.; et al. Shiga Toxin-Producing E. coli Infections Associated with Flour. N. Engl. J. Med. 2017, 377, 2036–2043. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A. Host and pathogen determinants of verocytotoxin-producing Escherichia coli-associated hemolytic uremic syndrome. Kidney Int. 2009, 75, S4–S7. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A. Emerging Public Health Challenges of Shiga Toxin-Producing Escherichia coli Related to Changes in the Pathogen, the Population, and the Environment. Clin. Infect. Dis. 2017, 64, 371–376. [Google Scholar] [PubMed]
- Bowen, E.E.; Coward, R.J. Advances in our understanding of the pathogenesis of hemolytic uremic syndromes. Am. J. Physiol. Renal Physiol. 2018, 314, F454–F461. [Google Scholar] [CrossRef]
- Garg, A.X.; Suri, R.S.; Barrowman, N.; Rehman, F.; Matsell, D.; Rosas-Arellano, M.P.; Salvadori, M.; Haynes, R.B.; Clark, W.F. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: A systematic review, meta-analysis, and meta-regression. JAMA 2003, 290, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Rosales, A.; Hofer, J.; Zimmerhackl, L.-B.; Jungraithmayr, T.C.; Riedl, M.; Giner, T.; Strasak, A.; Orth-Höller, D.; Würzner, R.; Karch, H.; et al. Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin. Infect. Dis. 2012, 54, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Frankel, G.; Phillips, A.D. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: Getting off the pedestal. Cell. Microbiol. 2008, 10, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G. Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFU and actin pedestal assembly. FEBS J. 2010, 277, 2390–2402. [Google Scholar] [CrossRef] [PubMed]
- Farfan, M.J.; Torres, A.G. Molecular mechanisms that mediate colonization of Shiga toxin-producing Escherichia coli strains. Infect. Immun. 2012, 80, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Barth, H.; Schmidt, H. Toxins of Locus of Enterocyte Effacement-Negative Shiga Toxin-Producing Escherichia coli. Toxins 2018, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Ehrlenbach, S.; Brockmeyer, J.; Khan, A.B.; Huber, G.; Karch, H.; Sarg, B.; Lindner, H.; Würzner, R. EspP, a serine protease of enterohemorrhagic Escherichia coli, impairs complement activation by cleaving complement factors C3/C3b and C5. Infect. Immun. 2010, 78, 4294–4301. [Google Scholar] [CrossRef]
- Bauwens, A.; Betz, J.; Meisen, I.; Kemper, B.; Karch, H.; Müthing, J. Facing glycosphingolipid-Shiga toxin interaction: Dire straits for endothelial cells of the human vasculature. Cell. Mol. Life Sci. 2013, 70, 425–457. [Google Scholar] [CrossRef]
- Rutjes, N.W.P.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef]
- Gallegos, K.M.; Conrady, D.G.; Karve, S.S.; Gunasekera, T.S.; Herr, A.B.; Weiss, A.A. Shiga toxin binding to glycolipids and glycans. PLoS ONE 2012, 7, e30368. [Google Scholar] [CrossRef] [PubMed]
- Louise, C.B.; Obrig, T.G. Shiga toxin-associated hemolytic uremic syndrome: Combined cytotoxic effects of shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect. Immun. 1992, 60, 1536–1543. [Google Scholar] [PubMed]
- Clayton, F.; Pysher, T.J.; Lou, R.; Kohan, D.E.; Denkers, N.D.; Tesh, V.L.; Taylor, F.B.; Siegler, R.L. Lipopolysaccharide upregulates renal shiga toxin receptors in a primate model of hemolytic uremic syndrome. Am. J. Nephrol. 2005, 25, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ito, S.; Honda, M. Hemolytic uremic syndrome induced by lipopolysaccharide and Shiga-like toxin. Pediatr. Nephrol. 2004, 19, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Keepers, T.R.; Psotka, M.A.; Gross, L.K.; Obrig, T.G. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J. Am. Soc. Nephrol. JASN 2006, 17, 3404–3414. [Google Scholar] [CrossRef] [PubMed]
- Zanchi, C.; Zoja, C.; Morigi, M.; Valsecchi, F.; Liu, X.Y.; Rottoli, D.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Mapelli, P.; et al. Fractalkine and CX3CR1 mediate leukocyte capture by endothelium in response to Shiga toxin. J. Immunol. 2008, 181, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef]
- O’Brien, A.D.; Tesh, V.L.; Donohue-Rolfe, A.; Jackson, M.P.; Olsnes, S.; Sandvig, K.; Lindberg, A.A.; Keusch, G.T. Shiga toxin: Biochemistry, genetics, mode of action, and role in pathogenesis. Curr. Top. Microbiol. Immunol. 1992, 180, 65–94. [Google Scholar]
- Lingwood, C.A. Role of verotoxin receptors in pathogenesis. Trends Microbiol. 1996, 4, 147–153. [Google Scholar] [CrossRef]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello-Pellegrini, T.N.; Moslemi-Naeini, M.; Marsden, P.A. New insights into Shiga toxin-mediated endothelial dysfunction in hemolytic uremic syndrome. Virulence 2013, 4, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 2012, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ardissino, G.; Patfield, S.; Cheng, L.W.; Silva, C.J.; Brigotti, M. An Improved Method for the Sensitive Detection of Shiga Toxin 2 in Human Serum. Toxins 2018, 10, 59. [Google Scholar] [CrossRef]
- Obrig, T.G.; Karpman, D. Shiga toxin pathogenesis: Kidney complications and renal failure. Curr. Top. Microbiol. Immunol. 2012, 357, 105–136. [Google Scholar] [PubMed]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E.; et al. Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef]
- Ståhl, A.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.-C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef]
- Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins 2017, 9, 376. [Google Scholar] [CrossRef]
- Morigi, M.; Micheletti, G.; Figliuzzi, M.; Imberti, B.; Karmali, M.A.; Remuzzi, A.; Remuzzi, G.; Zoja, C. Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood 1995, 86, 4553–4558. [Google Scholar] [PubMed]
- Zoja, C.; Angioletti, S.; Donadelli, R.; Zanchi, C.; Tomasoni, S.; Binda, E.; Imberti, B.; te Loo, M.; Monnens, L.; Remuzzi, G.; et al. Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-kappaB dependent up-regulation of IL-8 and MCP-1. Kidney Int. 2002, 62, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Matussek, A.; Lauber, J.; Bergau, A.; Hansen, W.; Rohde, M.; Dittmar, K.E.J.; Gunzer, M.; Mengel, M.; Gatzlaff, P.; Hartmann, M.; et al. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 2003, 102, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Galbusera, M.; Binda, E.; Imberti, B.; Gastoldi, S.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood 2001, 98, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Lo, N.C.; Turner, N.A.; Cruz, M.A.; Moake, J. Interaction of Shiga toxin with the A-domains and multimers of von Willebrand Factor. J. Biol. Chem. 2013, 288, 33118–33123. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello-Pellegrini, T.N.; Yuen, D.A.; Page, A.V.; Patel, S.; Soltyk, A.M.; Matouk, C.C.; Wong, D.K.; Turgeon, P.J.; Fish, J.E.; Ho, J.J.D.; et al. The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J. Clin. Investig. 2012, 122, 759–776. [Google Scholar] [CrossRef] [PubMed]
- Patry, C.; Betzen, C.; Fathalizadeh, F.; Fichtner, A.; Westhoff, J.H.; Fleming, T.; Eckstein, V.; Bruckner, T.; Bielaszewska, M.; Karch, H.; et al. Endothelial progenitor cells accelerate endothelial regeneration in an in vitro model of Shigatoxin-2a-induced injury via soluble growth factors. Am. J. Physiol. Renal Physiol. 2018, 315, F861–F869. [Google Scholar] [CrossRef]
- Morigi, M.; Buelli, S.; Zanchi, C.; Longaretti, L.; Macconi, D.; Benigni, A.; Moioli, D.; Remuzzi, G.; Zoja, C. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am. J. Pathol. 2006, 169, 1965–1975. [Google Scholar] [CrossRef]
- Hughes, A.K.; Stricklett, P.K.; Schmid, D.; Kohan, D.E. Cytotoxic effect of Shiga toxin-1 on human glomerular epithelial cells. Kidney Int. 2000, 57, 2350–2359. [Google Scholar] [CrossRef]
- Ergonul, Z.; Clayton, F.; Fogo, A.B.; Kohan, D.E. Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr. Nephrol. 2003, 18, 246–253. [Google Scholar]
- Dettmar, A.K.; Binder, E.; Greiner, F.R.; Liebau, M.C.; Kurschat, C.E.; Jungraithmayr, T.C.; Saleem, M.A.; Schmitt, C.-P.; Feifel, E.; Orth-Höller, D.; et al. Protection of human podocytes from shiga toxin 2-induced phosphorylation of mitogen-activated protein kinases and apoptosis by human serum amyloid P component. Infect. Immun. 2014, 82, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.K.; Stricklett, P.K.; Kohan, D.E. Shiga toxin-1 regulation of cytokine production by human glomerular epithelial cells. Nephron 2001, 88, 14–23. [Google Scholar] [CrossRef]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin triggers endothelial and podocyte injury: The role of complement activation. Pediatr. Nephrol. 2017, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Remuzzi, G.; Noris, M. Hemolytic uremic syndrome. Semin. Immunopathol. 2014, 36, 399–420. [Google Scholar] [CrossRef]
- Orth-Höller, D.; Würzner, R. Role of complement in enterohemorrhagic Escherichia coli-Induced hemolytic uremic syndrome. Semin. Thromb. Hemost. 2014, 40, 503–507. [Google Scholar]
- Keir, L.S.; Saleem, M.A. Current evidence for the role of complement in the pathogenesis of Shiga toxin haemolytic uraemic syndrome. Pediatr. Nephrol. 2014, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Lambris, J.D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Monnens, L.; Molenaar, J.; Lambert, P.H.; Proesmans, W.; van Munster, P. The complement system in hemolytic-uremic syndrome in childhood. Clin. Nephrol. 1980, 13, 168–171. [Google Scholar]
- Koster, F.T.; Boonpucknavig, V.; Sujaho, S.; Gilman, R.H.; Rahaman, M.M. Renal histopathology in the hemolytic-uremic syndrome following shigellosis. Clin. Nephrol. 1984, 21, 126–133. [Google Scholar] [PubMed]
- Robson, W.L.; Leung, A.K.; Fick, G.H.; McKenna, A.I. Hypocomplementemia and leukocytosis in diarrhea-associated hemolytic uremic syndrome. Nephron 1992, 62, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef]
- Thurman, J.M.; Marians, R.; Emlen, W.; Wood, S.; Smith, C.; Akana, H.; Holers, V.M.; Lesser, M.; Kline, M.; Hoffman, C.; et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 2009, 4, 1920–1924. [Google Scholar] [CrossRef]
- Ferraris, J.R.; Ferraris, V.; Acquier, A.B.; Sorroche, P.B.; Saez, M.S.; Ginaca, A.; Mendez, C.F. Activation of the alternative pathway of complement during the acute phase of typical haemolytic uraemic syndrome. Clin. Exp. Immunol. 2015, 181, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Rebetz, J.; Loos, S.; Herthelius, M.; Kristoffersson, A.-C.; Englund, E.; Chromek, M.; Karpman, D. Early Terminal Complement Blockade and C6 Deficiency Are Protective in Enterohemorrhagic Escherichia coli-Infected Mice. J. Immunol. 2016, 197, 1276–1286. [Google Scholar] [CrossRef]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga toxin activates complement and binds factor H: Evidence for an active role of complement in hemolytic uremic syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef]
- Poolpol, K.; Orth-Höller, D.; Speth, C.; Zipfel, P.F.; Skerka, C.; de Córdoba, S.R.; Brockmeyer, J.; Bielaszewska, M.; Würzner, R. Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol. Immunol. 2014, 58, 77–84. [Google Scholar] [CrossRef]
- Ehrlenbach, S.; Rosales, A.; Posch, W.; Wilflingseder, D.; Hermann, M.; Brockmeyer, J.; Karch, H.; Satchell, S.C.; Würzner, R.; Orth-Höller, D. Shiga toxin 2 reduces complement inhibitor CD59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect. Immun. 2013, 81, 2678–2685. [Google Scholar] [CrossRef]
- Arvidsson, I.; Ståhl, A.-L.; Hedström, M.M.; Kristoffersson, A.-C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef]
- Sims, P.J.; Faioni, E.M.; Wiedmer, T.; Shattil, S.J. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J. Biol. Chem. 1988, 263, 18205–18212. [Google Scholar] [PubMed]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J. Am. Soc. Nephrol. 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, D.; Kanno, K.; Takahashi, M.; Endo, Y.; Matsushita, M.; Fujita, T. The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. J. Immunol. 2011, 187, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M.; Kang, Y.; Tan, Y.S.; Pavlov, V.I.; Liu, B.; Boyle, D.C.; Kushak, R.I.; Skjoedt, M.-O.; Grabowski, E.F.; Taira, Y.; et al. Human mannose-binding lectin inhibitor prevents Shiga toxin-induced renal injury. Kidney Int. 2016, 90, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Crúz, M.A.; Zhang, H.; López, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef]
- Zoja, C.; Locatelli, M.; Pagani, C.; Corna, D.; Zanchi, C.; Isermann, B.; Remuzzi, G.; Conway, E.M.; Noris, M. Lack of the lectin-like domain of thrombomodulin worsens Shiga toxin-associated hemolytic uremic syndrome in mice. J. Immunol. 2012, 189, 3661–3668. [Google Scholar] [CrossRef]
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar] [CrossRef]
- Bettoni, S.; Galbusera, M.; Gastoldi, S.; Donadelli, R.; Tentori, C.; Spartà, G.; Bresin, E.; Mele, C.; Alberti, M.; Tortajada, A.; et al. Interaction between Multimeric von Willebrand Factor and Complement: A Fresh Look to the Pathophysiology of Microvascular Thrombosis. J. Immunol. 2017, 199, 1021–1040. [Google Scholar] [CrossRef]
- Albrecht, E.A.; Chinnaiyan, A.M.; Varambally, S.; Kumar-Sinha, C.; Barrette, T.R.; Sarma, J.V.; Ward, P.A. C5a-induced gene expression in human umbilical vein endothelial cells. Am. J. Pathol. 2004, 164, 849–859. [Google Scholar] [CrossRef]
- Monsinjon, T.; Gasque, P.; Chan, P.; Ischenko, A.; Brady, J.J.; Fontaine, M.C. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003, 17, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Schraufstatter, I.U.; Trieu, K.; Sikora, L.; Sriramarao, P.; DiScipio, R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J. Immunol. 2002, 169, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.; Pausa, M.; Nardon, E.; Introna, M.; Mantovani, A.; Dobrina, A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 1997, 185, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Polley, M.J.; Nachman, R.L. Human platelet activation by C3a and C3a des-arg. J. Exp. Med. 1983, 158, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, A.; Langer, H.F. Crosstalk between platelets and the complement system in immune protection and disease. Thromb. Haemost. 2013, 110, 910–919. [Google Scholar] [PubMed]
- Platt, J.L.; Vercellotti, G.M.; Lindman, B.J.; Oegema, T.R.; Bach, F.H.; Dalmasso, A.P. Release of heparan sulfate from endothelial cells. Implications for pathogenesis of hyperacute rejection. J. Exp. Med. 1990, 171, 1363–1368. [Google Scholar] [CrossRef]
- Saadi, S.; Platt, J.L. Transient perturbation of endothelial integrity induced by natural antibodies and complement. J. Exp. Med. 1995, 181, 21–31. [Google Scholar] [CrossRef]
- Spinale, J.M.; Ruebner, R.L.; Copelovitch, L.; Kaplan, B.S. Long-term outcomes of Shiga toxin hemolytic uremic syndrome. Pediatr. Nephrol. 2013, 28, 2097–2105. [Google Scholar] [CrossRef]
- Milford, D.V.; White, R.H.; Taylor, C.M. Prognostic significance of proteinuria one year after onset of diarrhea-associated hemolytic-uremic syndrome. J. Pediatr. 1991, 118, 191–194. [Google Scholar] [CrossRef]
- Repetto, H.A. Long-term course and mechanisms of progression of renal disease in hemolytic uremic syndrome. Kidney Int. 2005, 68, S102–S106. [Google Scholar] [CrossRef]
- Spizzirri, F.D.; Rahman, R.C.; Bibiloni, N.; Ruscasso, J.D.; Amoreo, O.R. Childhood hemolytic uremic syndrome in Argentina: Long-term follow-up and prognostic features. Pediatr. Nephrol. 1997, 11, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Goyal, M.; Kurnit, D.; Wharram, B.; Wiggins, J.; Holzman, L.; Kershaw, D.; Wiggins, R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001, 60, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Macconi, D.; Abbate, M.; Morigi, M.; Angioletti, S.; Mister, M.; Buelli, S.; Bonomelli, M.; Mundel, P.; Endlich, K.; Remuzzi, A.; et al. Permselective dysfunction of podocyte-podocyte contact upon angiotensin II unravels the molecular target for renoprotective intervention. Am. J. Pathol. 2006, 168, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Hodgin, J.B.; Bitzer, M.; Wickman, L.; Afshinnia, F.; Wang, S.Q.; O’Connor, C.; Yang, Y.; Meadowbrooke, C.; Chowdhury, M.; Kikuchi, M.; et al. Glomerular Aging and Focal Global Glomerulosclerosis: A Podometric Perspective. J. Am. Soc. Nephrol. 2015, 26, 3162–3178. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.B.; Shankland, S.J. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp. Nephrol. 2007, 106, e51–59. [Google Scholar] [CrossRef] [PubMed]
- Liapis, H.; Romagnani, P.; Anders, H.-J. New insights into the pathology of podocyte loss: Mitotic catastrophe. Am. J. Pathol. 2013, 183, 1364–1374. [Google Scholar] [CrossRef]
- Kaplan, B.S.; Trompeter, R.S.; Moake, J.L. Hemolytic Uremic Syndrome and Thrombotic Thrombocytopenic Purpura; Taylor & Francis: London, UK, 1992; ISBN 978-0-8247-8663-2. [Google Scholar]
- De Petris, L.; Patrick, J.; Christen, E.; Trachtman, H. Urinary podocyte mRNA excretion in children with D+HUS: A potential marker of long-term outcome. Ren. Fail. 2006, 28, 475–482. [Google Scholar] [CrossRef]
- Taylor, F.B.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the baboon responses to Shiga-like toxin: Descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Ochoa, F.; Oltra, G.; Gerhardt, E.; Hermes, R.; Cohen, L.; Damiano, A.E.; Ibarra, C.; Lago, N.R.; Zotta, E. Microalbuminuria and early renal response to lethal dose Shiga toxin type 2 in rats. Int. J. Nephrol. Renovasc. Dis. 2012, 5, 29–36. [Google Scholar] [PubMed]
- Morigi, M.; Locatelli, M.; Rota, C.; Buelli, S.; Corna, D.; Rizzo, P.; Abbate, M.; Conti, D.; Perico, L.; Longaretti, L.; et al. A previously unrecognized role of C3a in proteinuric progressive nephropathy. Sci. Rep. 2016, 6, 28445. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.R.; Johnson, S. Treatment and management of children with haemolytic uraemic syndrome. Arch. Dis. Child. 2018, 103, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef] [PubMed]
- Keenswijk, W.; Raes, A.; Vande Walle, J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur. J. Pediatr. 2018, 177, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.R.; Johnson, S. Eculizumab in the treatment of Shiga toxin haemolytic uraemic syndrome. Pediatr. Nephrol. 2018. [Google Scholar] [CrossRef]
- Lapeyraque, A.-L.; Malina, M.; Fremeaux-Bacchi, V.; Boppel, T.; Kirschfink, M.; Oualha, M.; Proulx, F.; Clermont, M.-J.; Le Deist, F.; Niaudet, P.; et al. Eculizumab in severe Shiga-toxin-associated HUS. N. Engl. J. Med. 2011, 364, 2561–2563. [Google Scholar] [CrossRef]
- Menne, J.; Nitschke, M.; Stingele, R.; Abu-Tair, M.; Beneke, J.; Bramstedt, J.; Bremer, J.P.; Brunkhorst, R.; Busch, V.; Dengler, R.; et al. Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: Case-control study. BMJ 2012, 345, e4565. [Google Scholar] [CrossRef]
- Loos, S.; Ahlenstiel, T.; Kranz, B.; Staude, H.; Pape, L.; Härtel, C.; Vester, U.; Buchtala, L.; Benz, K.; Hoppe, B.; et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: Presentation and short-term outcome in children. Clin. Infect. Dis. 2012, 55, 753–759. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Beutel, G.; Fleig, S.; Steinhoff, J.; Meyer, T.N.; Hafer, C.; Kuhlmann, U.; Bramstedt, J.; Panzer, U.; Vischedyk, M.; et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: An analysis of the German STEC-HUS registry. Nephrol. Dial. Transplant. 2012, 27, 3807–3815. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Remuzzi, G. Thrombotic microangiopathy: E. coli O104:H4 German outbreak: A missed opportunity. Nat. Rev. Nephrol. 2012, 8, 558–560. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buelli, S.; Zoja, C.; Remuzzi, G.; Morigi, M. Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Microorganisms 2019, 7, 15. https://doi.org/10.3390/microorganisms7010015
Buelli S, Zoja C, Remuzzi G, Morigi M. Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Microorganisms. 2019; 7(1):15. https://doi.org/10.3390/microorganisms7010015
Chicago/Turabian StyleBuelli, Simona, Carlamaria Zoja, Giuseppe Remuzzi, and Marina Morigi. 2019. "Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome" Microorganisms 7, no. 1: 15. https://doi.org/10.3390/microorganisms7010015
APA StyleBuelli, S., Zoja, C., Remuzzi, G., & Morigi, M. (2019). Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Microorganisms, 7(1), 15. https://doi.org/10.3390/microorganisms7010015