What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases

 ,
,  ,
, 

Abstract
1. Introduction
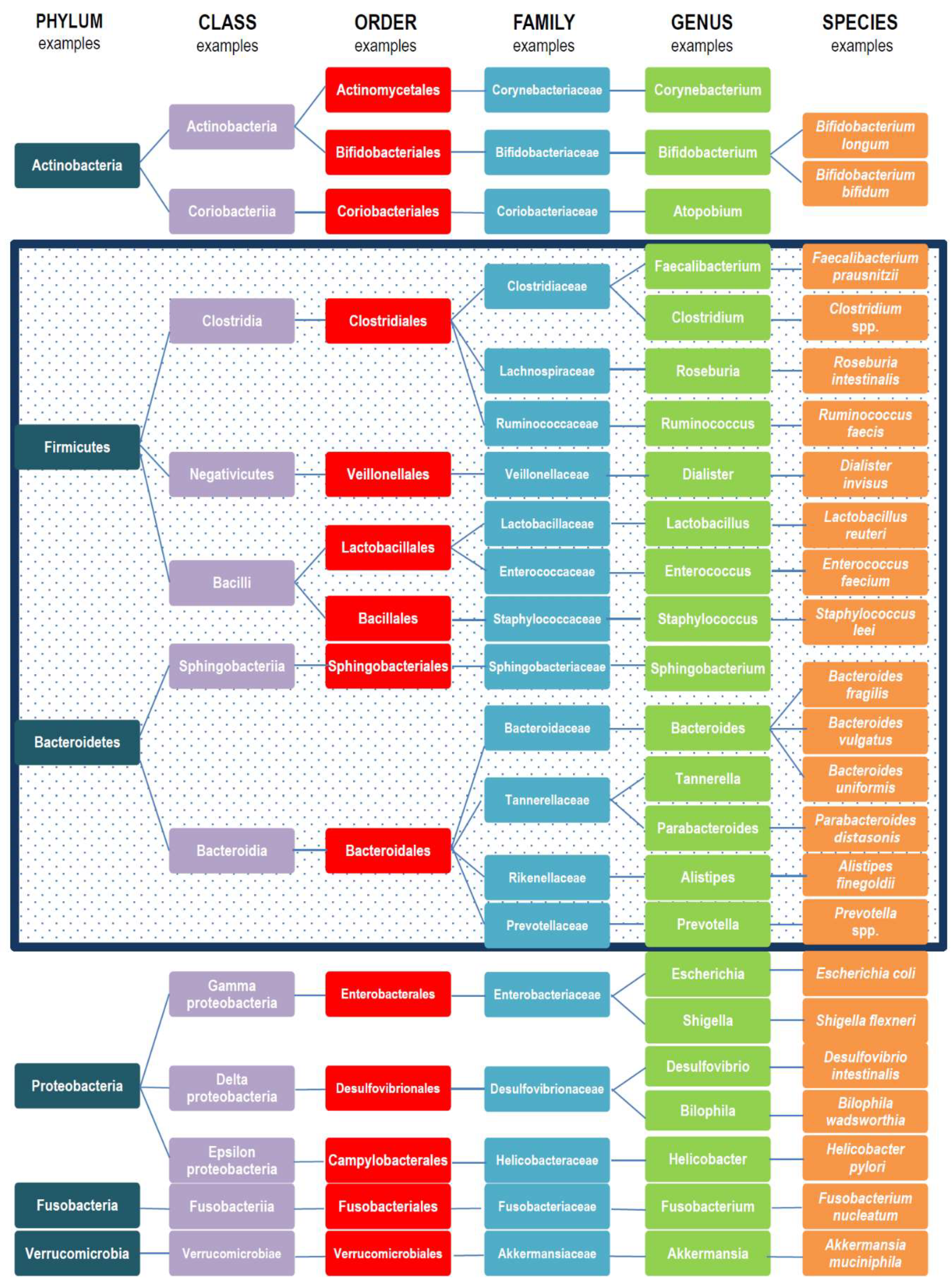
2. Gut Microbiota Variations
2.1. Variations in the Same Individual
2.1.1. Intestine Anatomical Regions
2.1.2. Infant Transitions
Birth Gestational Age
Type of Delivery
Methods of Milk Feeding
Weaning Period
2.1.3. Age
2.1.4. Antibiotics
2.2. Gut Microbiota Variations between Individuals
2.2.1. Enterotypes
2.2.2. Body Mass Index (BMI) Classes
2.2.3. Ethnicity, Dietary Habits, and Cultural Habits
2.2.4. Exercise Frequency
3. Gut Microbiota Variations in Health and Disease
3.1. Intestinal Disorders
3.1.1. Irritable Bowel Syndrome
3.1.2. Inflammatory Bowel Disease (IBD)
3.1.3. Celiac Disease
3.1.4. Colorectal Cancer (CRC)
3.2. Extra-Intestinal Disorders
3.2.1. Metabolic Disorders
Obesity
Type 2 Diabetes (T2D)
3.2.2. Central Nervous System (CNS)-Related Disorders
Alzheimer’s and Parkinson’s Diseases
Hepatic Encephalopathy
Autism Spectrum Disorders
Stress
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.E.; Holdeman, L.V. Human fecal flora: The normal flora of 20 Japanese-Hawaiians. Appl. Microbiol. 1974, 27, 961–979. [Google Scholar] [PubMed]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi-Man, O.; Davenport, E.R.; Gilad, Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: Evaluation of effective study designs. PLoS ONE 2013, 8, e53608. [Google Scholar] [CrossRef]
- Human Microbiome Jumpstart Reference Strains Consortium; Nelson, K.E.; Weinstock, G.M.; Highlander, S.K.; Worley, K.C.; Creasy, H.H.; Wortman, J.R.; Rusch, D.B.; Mitreva, M.; Sodergren, E.; et al. A catalog of reference genomes from the human microbiome. Science 2010, 328, 994–999. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Luckey, T.D. Introduction to intestinal microecology. Am. J. Clin. Nutr. 1972, 25, 1292–1294. [Google Scholar] [CrossRef]
- Khosravi, A.; Mazmanian, S.K. Disruption of the gut microbiome as a risk factor for microbial infections. Curr. Opin. Microbiol. 2013, 16, 221–227. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Laterza, L.; Rizzatti, G.; Gaetani, E.; Chiusolo, P.; Gasbarrini, A. The gut microbiota and immune system relationship in human graft-versus-host disease. Mediterr. J. Hematol. Infect. Dis. 2016, 8, e2016025. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Arboleya, S.; Binetti, A.; Salazar, N.; Fernández, N.; Solís, G.; Hernández-Barranco, A.; Margolles, A.; de Los Reyes-Gavilán, C.G.; Gueimonde, M. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol. Ecol. 2012, 79, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Hui, Y.; Obelitz-Ryom, K.; Brandt, A.B.; Kot, W.; Nielsen, D.S.; Thymann, T.; Sangild, P.T.; Nguyen, D.N. Neonatal gut and immune maturation is determined more by postnatal age than by post-conceptional age in moderately preterm pigs. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Butel, M.J.; Suau, A.; Campeotto, F.; Magne, F.; Aires, J.; Ferraris, L.; Kalach, N.; Leroux, B.; Dupont, C. Conditions of bifidobacterial colonization in preterm infants: A prospective analysis. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 577–582. [Google Scholar] [CrossRef]
- Gabrielli, O.; Zampini, L.; Galeazzi, T.; Padella, L.; Santoro, L.; Peila, C.; Giuliani, F.; Bertino, E.; Fabris, C.; Coppa, G.V. Preterm milk oligosaccharides during the first month of lactation. Pediatrics 2011, 128, e1520–e1531. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.A.; Gaerlan, S.; De Leoz, M.L.; Dimapasoc, L.; Kalanetra, K.M.; Lemay, D.G.; German, J.B.; Mills, D.A.; Lebrilla, C.B. Human milk oligosaccharides in premature infants: Absorption, excretion, and influence on the intestinal microbiota. Pediatr. Res. 2015, 78, 670–677. [Google Scholar] [CrossRef]
- Praticò, G.; Capuani, G.; Tomassini, A.; Baldassarre, M.E.; Delfini, M.; Miccheli, A. Exploring human breast milk composition by NMR-based metabolomics. Nat. Prod. Res. 2014, 28, 95–101. [Google Scholar] [CrossRef]
- Bering, S.B. Human milk oligosaccharides to prevent gut dysfunction and necrotizing enterocolitis in preterm neonates. Nutrients 2018, 10, 1461. [Google Scholar] [CrossRef] [PubMed]
- Mastromarino, P.; Capobianco, D.; Campagna, G.; Laforgia, N.; Drimaco, P.; Dileone, A.; Baldassarre, M.E. Correlation between lactoferrin and beneficial microbiota in breast milk and infant’s feces. Biometals 2014, 27, 1077–1086. [Google Scholar] [CrossRef]
- Salminen, S.; Gibson, G.R.; McCartney, A.L.; Isolauri, E. Influence of mode of delivery on gut microbiota composition in seven year old children. Gut 2004, 53, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [PubMed]
- Biasucci, G.; Benenati, B.; Morelli, L.; Bessi, E.; Boehm, G. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J. Nutr. 2008, 138, 1796S–1800S. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends Mol. Med. 2015, 21, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Pantoja-Feliciano, I.G.; Clemente, J.C.; Costello, E.K.; Perez, M.E.; Blaser, M.J.; Knight, R.; Dominguez-Bello, M.G. Biphasic assembly of the murine intestinal microbiota during early development. ISME J. 2013, 7, 1112–1115. [Google Scholar] [CrossRef]
- Grönlund, M.M.; Lehtonen, O.P.; Eerola, E.; Kero, P. Fecal microflora in healthy infants born by different methods of delivery: Permanent changes in intestinal flora after cesarean delivery. J. Pediatr. Gastroenterol. Nutr. 1999, 28, 19–25. [Google Scholar] [CrossRef]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Chari, R.S.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L. Gut microbiota of healthy Canadian infants: Profiles by mode of delivery and infant diet at 4 months. CMAJ 2013, 185, 385–394. [Google Scholar] [CrossRef]
- Sevelsted, A.; Stokholm, J.; Bønnelykke, K.; Bisgaard, H. Cesarean section and chronic immune disorders. Pediatrics 2015, 135, e92–e98. [Google Scholar] [CrossRef]
- Chen, G.; Chiang, W.L.; Shu, B.C.; Guo, Y.L.; Chiou, S.T.; Chiang, T.L. Associations of caesarean delivery and the occurrence of neurodevelopmental disorders, asthma or obesity in childhood based on Taiwan birth cohort study. BMJ Open 2017, 7, e017086. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; van den Brandt, P.A.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Bezirtzoglou, E.; Tsiotsias, A.; Welling, G.W. Microbiota profile in feces of breast- and formula-fed newborns by using fluorescence in situ hybridization (FISH). Anaerobe 2011, 17, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.C.; Costabile, A.; Holland, D.T.; Hoyles, L.; McCartney, A.L. Examination of faecal Bifidobacterium populations in breast- and formula-fed infants during the first 18 months of life. Microbiology 2010, 156, 3329–3341. [Google Scholar] [CrossRef]
- Marcobal, A.; Barboza, M.; Froehlich, J.W.; Block, D.E.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Consumption of human milk oligosaccharides by gut-related microbes. J. Agric. Food Chem. 2010, 58, 5334–5340. [Google Scholar] [CrossRef] [PubMed]
- Sakurama, H.; Kiyohara, M.; Wada, J.; Honda, Y.; Yamaguchi, M.; Fukiya, S.; Yokota, A.; Ashida, H.; Kumagai, H.; Kitaoka, M.; et al. Lacto-N-biosidase encoded by a novel gene of Bifidobacterium longum subspecies longum shows unique substrate specificity and requires a designated chaperone for its active expression. J. Biol. Chem. 2013, 288, 25194–25206. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Yahagi, K.; Mori, H.; Matsumoto, H.; Hara, T.; Tajima, S.; Ogawa, E.; Kodama, H.; Yamamoto, K.; Yamada, T.; et al. A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nat. Commun. 2016, 7, 11939. [Google Scholar] [CrossRef] [PubMed]
- Yaron, S.; Shachar, D.; Abramas, L.; Riskin, A.; Bader, D.; Litmanovitz, I.; Bar-Yoseph, F.; Cohen, T.; Levi, L.; Lifshitz, Y.; et al. Effect of high β-palmitate content in infant formula on the intestinal microbiota of term infants. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 376–381. [Google Scholar] [CrossRef]
- Mastromarino, P.; Capobianco, D.; Miccheli, A.; Praticò, G.; Campagna, G.; Laforgia, N.; Capursi, T.; Baldassarre, M.E. Administration of a multistrain probiotic product (VSL#3) to women in the perinatal period differentially affects breast milk beneficial microbiota in relation to mode of delivery. Pharmacol. Res. 2015, 95, 63–70. [Google Scholar] [CrossRef]
- Fallani, M.; Amarri, S.; Uusijarvi, A.; Adam, R.; Khanna, S.; Aguilera, M.; Gil, A.; Vieites, J.M.; Norin, E.; Young, D.; et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology 2011, 157, 1385–1392. [Google Scholar] [CrossRef]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [Google Scholar] [CrossRef]
- Tidjani Alou, M.; Lagier, J.C.; Raoult, D. Diet influence on the gut microbiota and dysbiosis related to nutritional disorders. Hum. Microbiome J. 2016, 1, 3–11. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Toshitaka, O.; Kumiko, K.; Hirosuke, S.; Nanami, H.; Sachiko, T.; Jin-Zhong, X.; Fumiaki, A.; Ro, O. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar]
- Guigoz, Y.; Doré, J.; Schiffrin, E.J. The inflammatory status of old age can be nurtured from the intestinal environment. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cobas, A.E.; Artacho, A.; Knecht, H.; Ferrús, M.L.; Friedrichs, A.; Ott, S.J.; Moya, A.; Latorre, A.; Gosalbes, M.J. Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS ONE 2013, 8, e80201. [Google Scholar] [CrossRef] [PubMed]
- Iizumi, T.; Battaglia, T.; Ruiz, V.; Perez, G.I. Gut microbiome and antibiotics. Arch. Med. Res. 2017, 48, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Bai, J.; Hu, Y.; Bruner, D.W. Composition of gut microbiota and its association with body mass index and lifestyle factors in a cohort of 7–18 years old children from the American Gut Project. Pediatr. Obes. 2018. [Google Scholar] [CrossRef]
- Karlsson, C.L.; Onnerfält, J.; Xu, J.; Molin, G.; Ahrné, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Kim, H.N.; Kim, S.E.; Heo, S.G.; Chang, Y.; Ryu, S.; Shin, H.; Kim, H.L. Comparative analysis of gut microbiota associated with body mass index in a large Korean cohort. BMC Microbiol. 2017, 17, 151. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, L.; Van Hoorenbeeck, K.; Kortleven, I.; Van Noten, C.; Hens, N.; Vael, C.; Goossens, H.; Desager, K.N.; Vankerckhoven, V. Differences in gut microbiota composition between obese and lean children: A cross-sectional study. Gut Pathog. 2013, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Borgo, F.; Lassandro, C.; Verduci, E.; Morace, G.; Borghi, E.; Berry, D. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Environ. Microbiol. 2017, 19, 95–105. [Google Scholar] [CrossRef]
- Borgo, F.; Riva, A.; Benetti, A.; Casiraghi, M.C.; Bertelli, S.; Garbossa, S.; Anselmetti, S.; Scarone, S.; Pontiroli, A.E.; Morace, G.; et al. Microbiota in anorexia nervosa: The triangle between bacterial species, metabolites and psychological tests. PLoS ONE 2017, 12, e0179739. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise modifies the gut microbiota with positive health effects. Oxid. Med. Cell. Longev. 2017, 3831972. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, Y.; Muniz Pedrogo, D.A.; Kashyap, P.C. Irritable bowel syndrome: A gut microbiota-related disorder? Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Carroll, I.M.; Chang, Y.H.; Park, J.; Sartor, R.B.; Ringel, Y. Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut Pathog. 2010, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Krogius-Kurikka, L.; Lyra, A.; Malinen, E.; Aarnikunnas, J.; Tuimala, J.; Paulin, L.; Mäkivuokko, H.; Kajander, K.; Palva, A. Microbial community analysis reveals high level phylogenetic alterations in the overall gastrointestinal microbiota of diarrhoea-predominant irritable bowel syndrome sufferers. BMC Gastroenterol. 2009, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; de Vos, W.M.; Palva, A. Gastrointestinal microbiota in irritable bowel syndrome: Present state and perspectives. Microbiology 2010, 156, 3205–3215. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Hansen, R.; Russell, R.K.; Reiff, C.; Louis, P.; McIntosh, F.; Berry, S.H.; Mukhopadhya, I.; Bisset, W.M.; Barclay, A.R.; Bishop, J.; et al. Microbiota of de-novo pediatric IBD: Increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am. J. Gastroenterol. 2012, 107, 1913–1922. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Marasco, G.; Di Biase, A.R.; Schiumerini, R.; Eusebi, L.H.; Iughetti, L.; Ravaioli, F.; Scaioli, E.; Colecchia, A.; Festi, D. Gut microbiota and celiac disease. Dig. Dis. Sci. 2016, 61, 1461–1472. [Google Scholar] [CrossRef]
- Chander, A.M.; Yadav, H.; Jain, S.; Bhadada, S.K.; Dhawan, D.K. Cross-talk between gluten, intestinal microbiota and intestinal mucosa in celiac disease: Recent advances and basis of autoimmunity. Front. Microbiol. 2018, 9, 2597. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Nadal, I.; Medina, M.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010, 10, 63. [Google Scholar] [CrossRef]
- Nadal, I.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in faecal and duodenal Bifidobacterium species composition inactive and non-active coeliac disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2009, 62, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef]
- Shen, X.J.; Rawls, J.F.; Randall, T.; Burcal, L.; Mpande, C.N.; Jenkins, N.; Jovov, B.; Abdo, Z.; Sandler, R.S.; Keku, T.O. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 2010, 1, 138–147. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Mele, M.C.; Merendino, N.; Cintoni, M.; Anselmi, G.; Caporossi, A.; Gasbarrini, A.; Minnella, A.M. The role of diet, micronutrients and the gut microbiota in age-related macular degeneration: New perspectives from the gut-retina axis. Nutrients 2018, 10, 1677. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Lazarevic, V.; Derrien, M.; Everard, A.; Van Roye, M.; Knauf, C.; Valet, P.; Girard, M.; Muccioli, G.G.; François, P. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: Impact on apelin regulation in adipose tissue. Front. Microbiol. 2011, 2, 149. [Google Scholar] [CrossRef]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Gut microbiota and obesity: Lessons from the microbiome. Brief. Funct. Genom. 2013, 12, 381–387. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. SME J. 2010, 4, 232–241. [Google Scholar] [CrossRef]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A common factor in human diseases. Biomed. Res. Int. 2017, 9351507. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Walsh, C.J.; Guinane, C.M.; O’Toole, P.W.; Cotter, P.D. Beneficial modulation of the gut microbiota. FEBS Lett. 2014, 588, 4120–4130. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Watanabe, K.; Kimura, I. Gut microbiota dysbiosis drives and implies novel therapeutic strategies for diabetes mellitus and related metabolic diseases. Front. Immunol. 2017, 8, 1882. [Google Scholar] [CrossRef]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed on 1 November 2018).
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Hopfner, F.; Künstner, A.; Müller, S.H.; Künzel, S.; Zeuner, K.E.; Margraf, N.G.; Deuschl, G.; Baines, J.F.; Kuhlenbäumer, G. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017, 1667, 41–45. [Google Scholar] [CrossRef]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. 2017, 32, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Ridlon, J.M.; Hylemon, P.B.; Thacker, L.R.; Heuman, D.M.; Smith, S.; Sikaroodi, M.; Gillevet, P.M. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M. Desulfovibrio species are potentially important in regressive autism. Med. Hypotheses 2011, 77, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Downes, J.; Summanen, P.H. Microbiology of regressive autism. Anaerobe 2012, 18, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, Y.; Dy, A.B.C.; Hagerman, R.J. The gut microbiota and autism spectrum disorders. Front. Cell. Neurosci. 2017, 11, 120. [Google Scholar] [CrossRef]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Low relative abundances of the mucolytic bacterium Akkermansia muciniphila and Bifidobacterium spp. in feces of children with autism. Appl. Environ. Microbiol. 2011, 77, 6718–6721. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.L.; Hornig, M.; Parekh, T.; Lipkin, W.I. Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. MBio 2012, 3, e00261-11. [Google Scholar] [CrossRef]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108, 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Cryan, J.F.; Fitzgerald, G.F.; Ross, R.P.; Dinan, T.G.; Stanton, C. Gut microbiota, the pharmabiotics they produce and host health. Proc. Nutr. Soc. 2014, 73, 477–489. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gut Microbiota Abundance | Bacteria Diversity | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Actinobacteria | Bacteroidetes | Firmicutes | Proteobacteria | Fusobacteria | Verrucomicrobia | Euryarchaeota | ||||
| Anatomical part of gut tract | Small intestine | Lactobacillus | Enterobacteriaceae * | [14] | ||||||
| Colon | Bacteroidaceae * Prevotellaceae * Rikenellaceae * | Lachnospiraceae * Ruminococcaceae * | ||||||||
| Gestational age | Preterm birth (<37 weeks of gestation) | Bifidobacterium spp.↓ Atopobium spp.↓ | Bacteroides *↓ (non-secretor mothers) | Firmicutes *↓ (non-secretor mothers) Lactobacillus↑ Ruminococcus spp. Lachnospiraceae * Peptostreptococcaceae * Clostridiaceae * | Enterobacteriaceae *↑ Enterococcus spp.↑ | ↓ | [15,16,19] | |||
| Full-term birth | Bifidobacterium spp.↑ | Bacteroidetes *↑ | Ruminococcus spp. Lachnospiraceae * Peptostreptococcaceae * Clostridiaceae * | Enterobacteriaceae * | ↑ | [16,20] | ||||
| Type of delivery | Vaginal delivery | Bifidobacterium spp.↑ Bifidobacterium longum↑ Bifidobacterium catenulatum↑ | Prevotella↑ Bacteroides fragilis↑ | Lactobacillus↑ Staphylococcus↑ Streptococcus ↑ | Escherichia↑ | Sneathia↑ | ↑ | [22,23,24,25,26] | ||
| C-section | Corynebacterium↑ Propionibacterium↑ | Bacteroides *↓ | Staphylococcus↑ | Escherichia↓ Shigella↓ | ↓ | [21,22,23,27] | ||||
| Methods of (milk) feedings | Breast milk | Bifidobacterium↑↑ | Lactobacillus↑ Staphylococcus ↑ | Enterococcus↑ | ↑ | [27,30,31,32,38] | ||||
| Artificial milk | Bifidobacterium↑ | Bacteroides↑ | Clostridium↑ Clostridium difficile↑ Lactobacillus↑ | Escherichia↑ | ↓ | [27,30,31,32] | ||||
| Introduction of solid food | Bifidobacterium↑ | Bacteroidetes *↑ Bacteroides↑ | Firmicutes *↑ Lactobacilli↑ Clostridium coccoides↑ | ↑ | [36,37] | |||||
| Human age | Childhood (first year of life) | Bifidobacterium | Bacteroides | Veillonella C. coccoides C. botulinum | Akkermansia muciniphila | ↑ | [38,39] | |||
| 2–3 years old to adult | Bifidobacteriaceae * Coriobacteriaceae * | Bacteroidaceae * Prevotellaceae * Rikenellaceae * | Lachnospiraceae Ruminococcaceae | Proteobacteria * | Fusobacteria * | Akkermansia muciniphila | Methanobrevibacter smithii | ↑ | [38] | |
| Over 70 | Bifidobacteriaceae↓ | Clostridium *↓ | Proteobacteria *↑ | ↓ | [40] | |||||
| Antibiotic treatments | Macrolide | Actinobacteria *↓ | Bacteroides↑ | Firmicutes *↓ | Proteobacteria *↑ | ↓ | [43] | |||
| Clarithromycin | Actinobacteria *↓ | Bacteroides↑ | Firmicutes *↓ | Proteobacteria *↑ | ↓ | |||||
| Vancomycin | Lactobacillus↓ Clostridium↓ | ↓ | ||||||||
| Ciprofloxacin | Bifidobacterium↓ | Alistipes↓ Bacteroides↑ | Faecalibacterium↓ Oscillospira↓ Ruminococcus↓ Dialister↓ | ↓ | ||||||
| Clindamycin | Bifidobacteriaceae↓ Lactobacillus↓ | ↓ | ||||||||
| Gut Microbiota Abundance | Bacteria Diversity | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Actinobacteria | Bacteroidetes | Firmicutes | Proteobacteria | Verrucomicrobia | Euryarchaeota | Spirochaetes | ||||
| Enterotype | I | Slackia | Bacteroides(1) Parabacteroides | Clostridiales * Alkaliphilus Lactobacillus Catenibacterium | Geobacter | Methanobrevibacter smithii | [13] | |||
| II | Eggerthella | Prevotella(1) | Veillonella Ruminococcaceae * Holdemania Peptostreptococcaceae * Staphylococcus Leuconostoc | Desulfovibrionaceae * Rhodospirillum Helicobacter Escherichia Shigella | Akkermansia muciniphila | |||||
| III | Gordonibacter | Sphingobacterium | Ruminococcus(1) Staphylococcus Marvinbryantia Symbiobacterium Ruminococcaceae * Dialister | Akkermansia muciniphila | ||||||
| BMI level (among children and adolescents) | Overweight to obese level | Bifidobacterium↑ B. vulgatus↓ | Bacteroidetes *↓ | Lactobacillus↑ Firmicutes *↑ | Proteobacteria *↑ | ↓ | [46,47,49,50] | |||
| Underweight level | Roseburia spp.↓ Ruminococcus↓ Clostridium spp.↓ | Enterobacteriaceae *↑ | Methanobrevibacter smithii↑ | ↓ | [46,51] | |||||
| Exercise frequency | High performance sport | Bacteroides↓ | Ruminococcaceae *↑ Lactobacillus↓ Lactobacillaceae *↓ | Succinivibrionaceae *↑ | Akkermansiaceae↓ | ↑ | [57] | |||
| Children and adolescents daily exercise | Clostridiales↑ Roseburia↑ Lachnospiraceae↑ Erysipelotrichaceae↑ | ↑ | [46] | |||||||
| Type of diet, communities, climate, and geographical sites | High-fiber diet (African diet) | Bifidobacterium↓ | Prevotella↑ Bacteroides↓ | Eubacterium↑ Oscillibacter↑ Butyricicoccus↑ Sporobacter↑ Blautia↓ Dorea↓ Lachnospiraceae *↓ Roseburia↓ Faecalibacterium↓ Ruminococcus↓ Erysipelotrichaceae *↓ | Succinivibrio↑ | Treponema↑ | ↑ | [53,55] | ||
| High-fat and animal protein diet (Western diet) | Actinobacteria *↑ | Bacteroides↑ Alistipes↑ Barnesiella↑ | Roseburia↓ Eubacterium rectale↓ Ruminococcus bromii↓ | Bilophila↑ | ↓ | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. https://doi.org/10.3390/microorganisms7010014
Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms. 2019; 7(1):14. https://doi.org/10.3390/microorganisms7010014
Chicago/Turabian StyleRinninella, Emanuele, Pauline Raoul, Marco Cintoni, Francesco Franceschi, Giacinto Abele Donato Miggiano, Antonio Gasbarrini, and Maria Cristina Mele. 2019. "What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases" Microorganisms 7, no. 1: 14. https://doi.org/10.3390/microorganisms7010014
APA StyleRinninella, E., Raoul, P., Cintoni, M., Franceschi, F., Miggiano, G. A. D., Gasbarrini, A., & Mele, M. C. (2019). What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms, 7(1), 14. https://doi.org/10.3390/microorganisms7010014