
A Comparison of Three Automated Nucleic Acid Extraction Systems for Human Stool Samples


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. DNA Extraction and Quantification
FastDNA Spin Kit for Soil (MP-Biomedical Catalog No: 116560200)
2.3. 16S Amplicon Sequencing Library Preparation and Analysis
3. Results
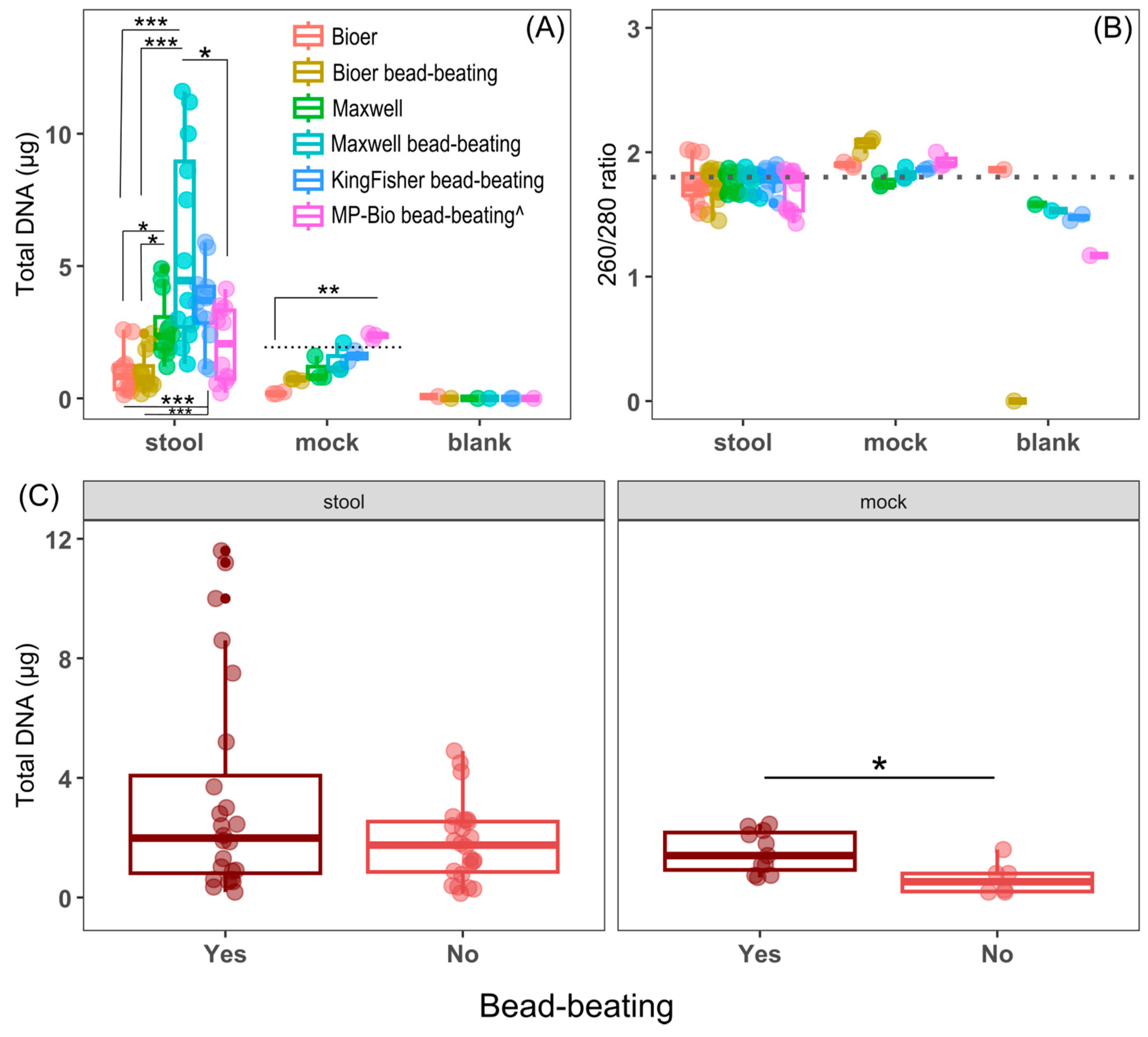
3.1. Comparison of DNA Yield and Purity of Fecal and Mock Microbiotas Between the DNA Extraction Methods
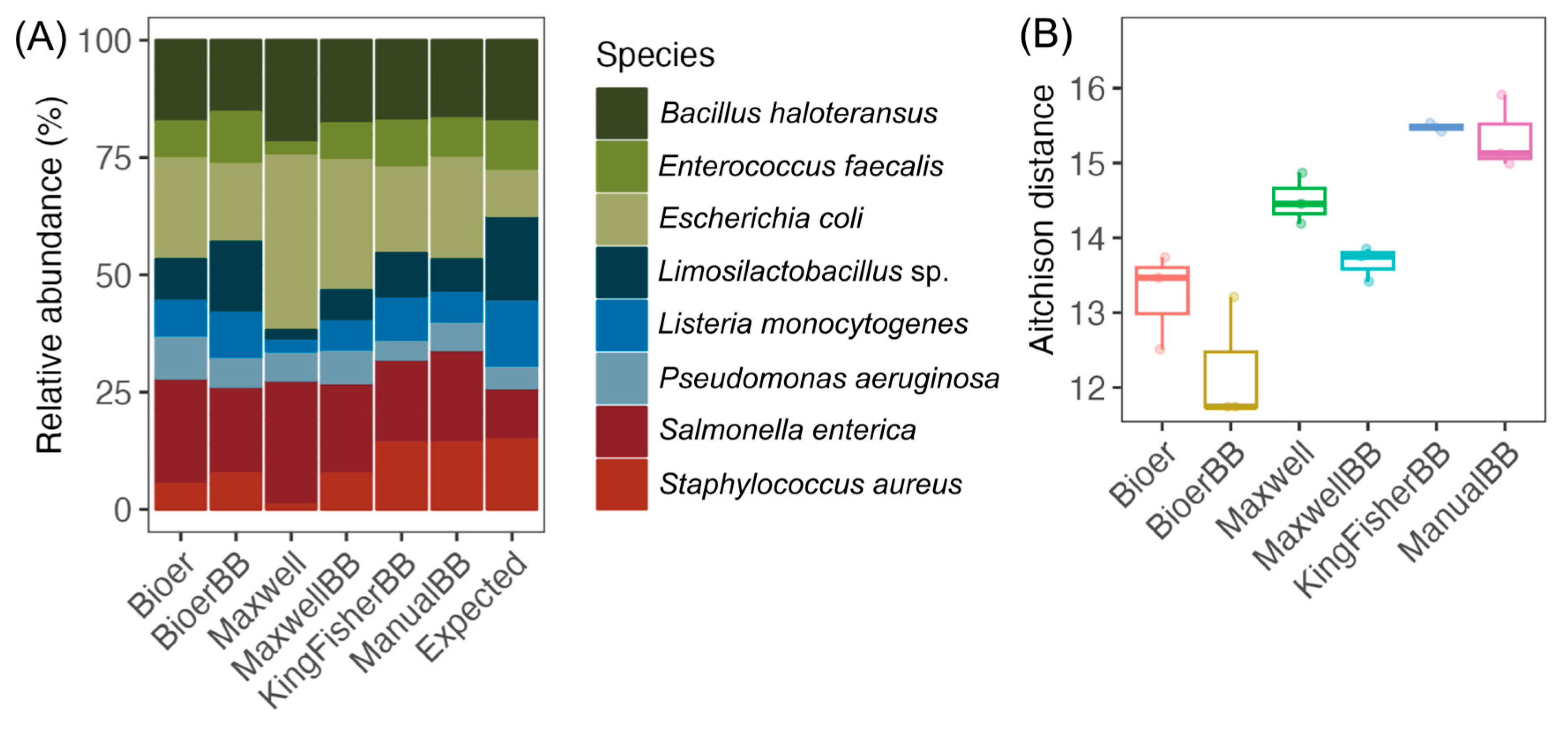
3.2. Differences in the Mock Community of Nucleic Acid Extractors
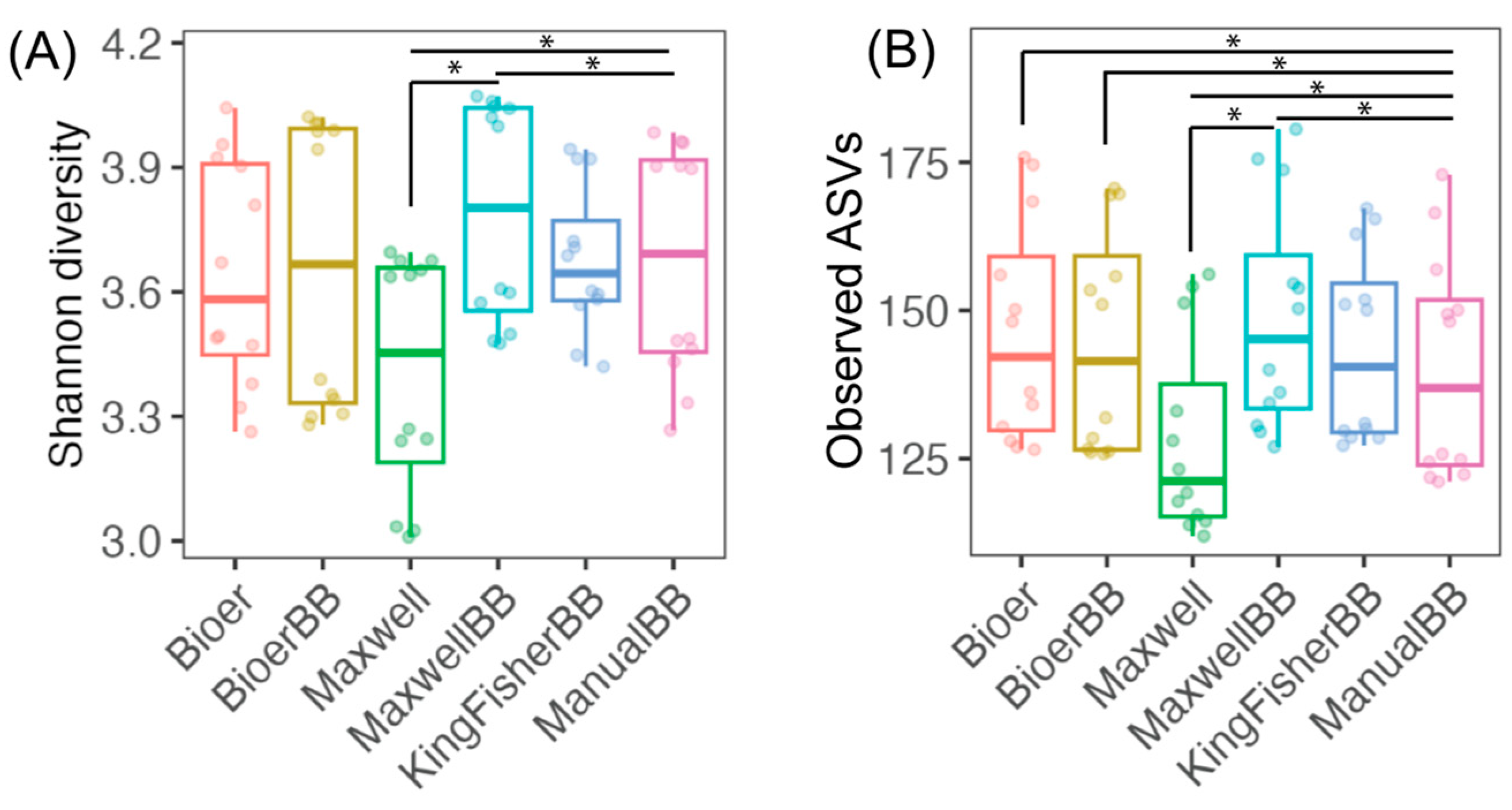
3.3. Alpha-Diversity of Fecal Microbiota Between Nucleic Acid Extractors
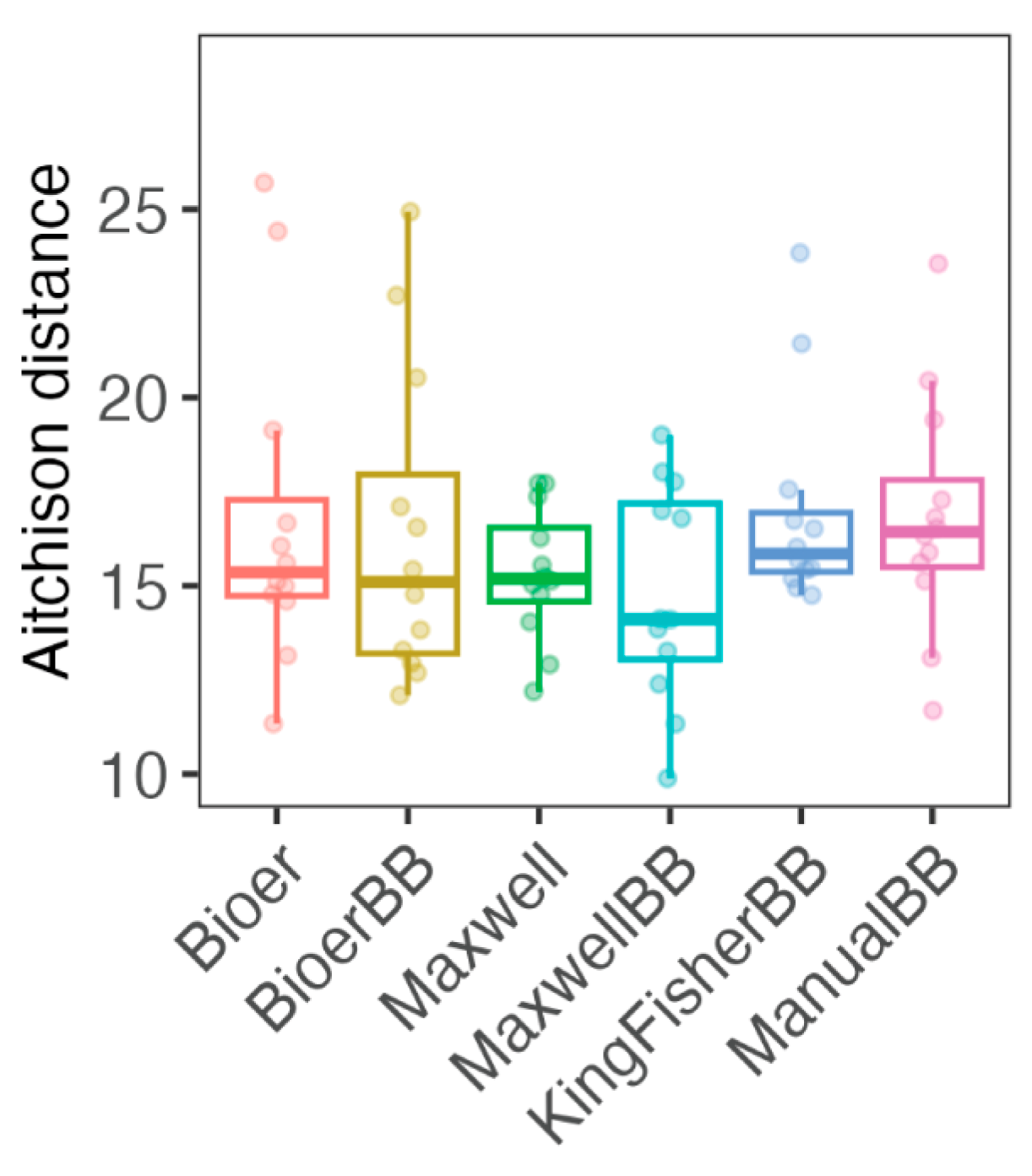
3.4. Variability of Microbiota Between Nucleic Acid Extraction Systems
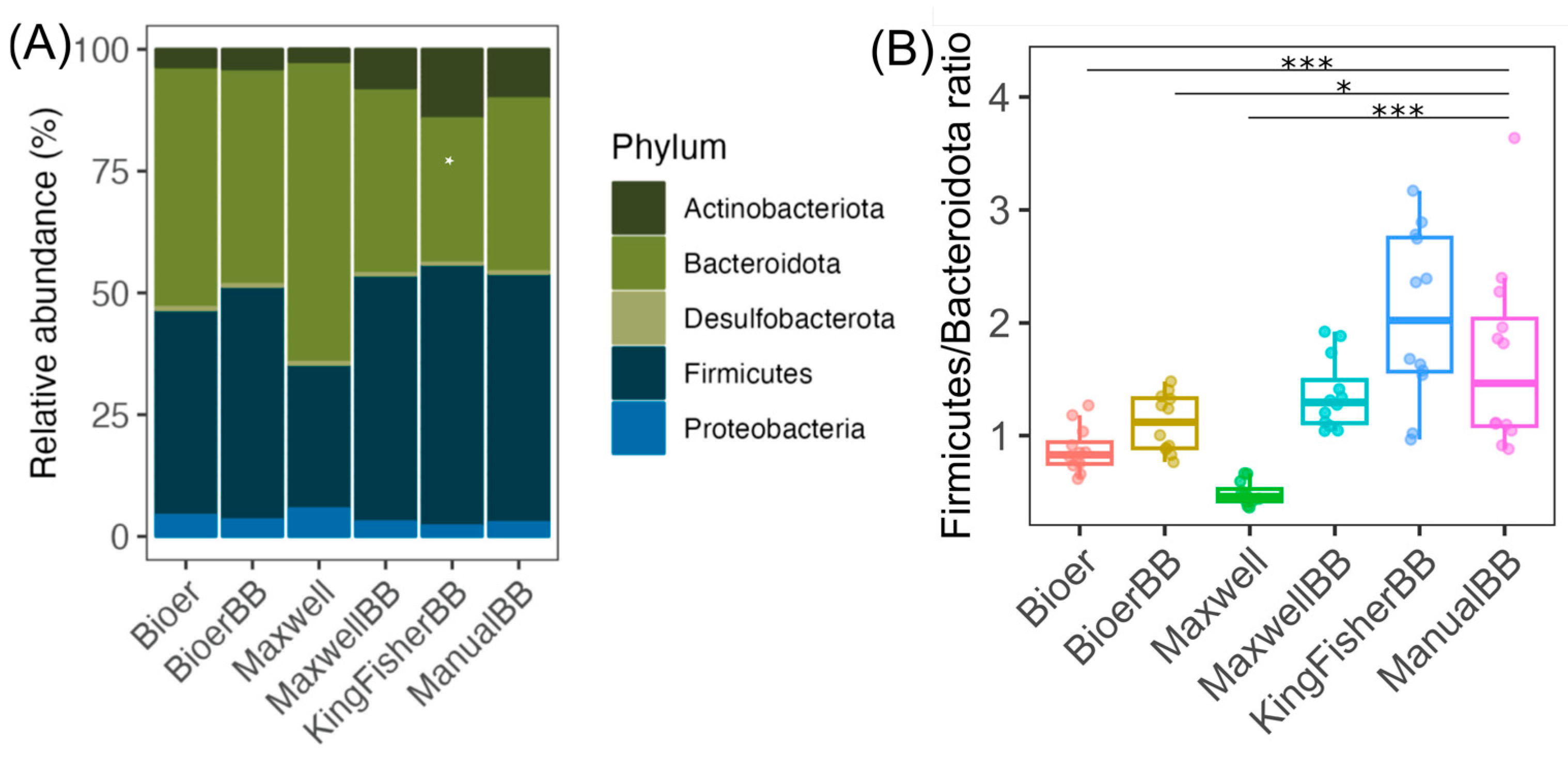
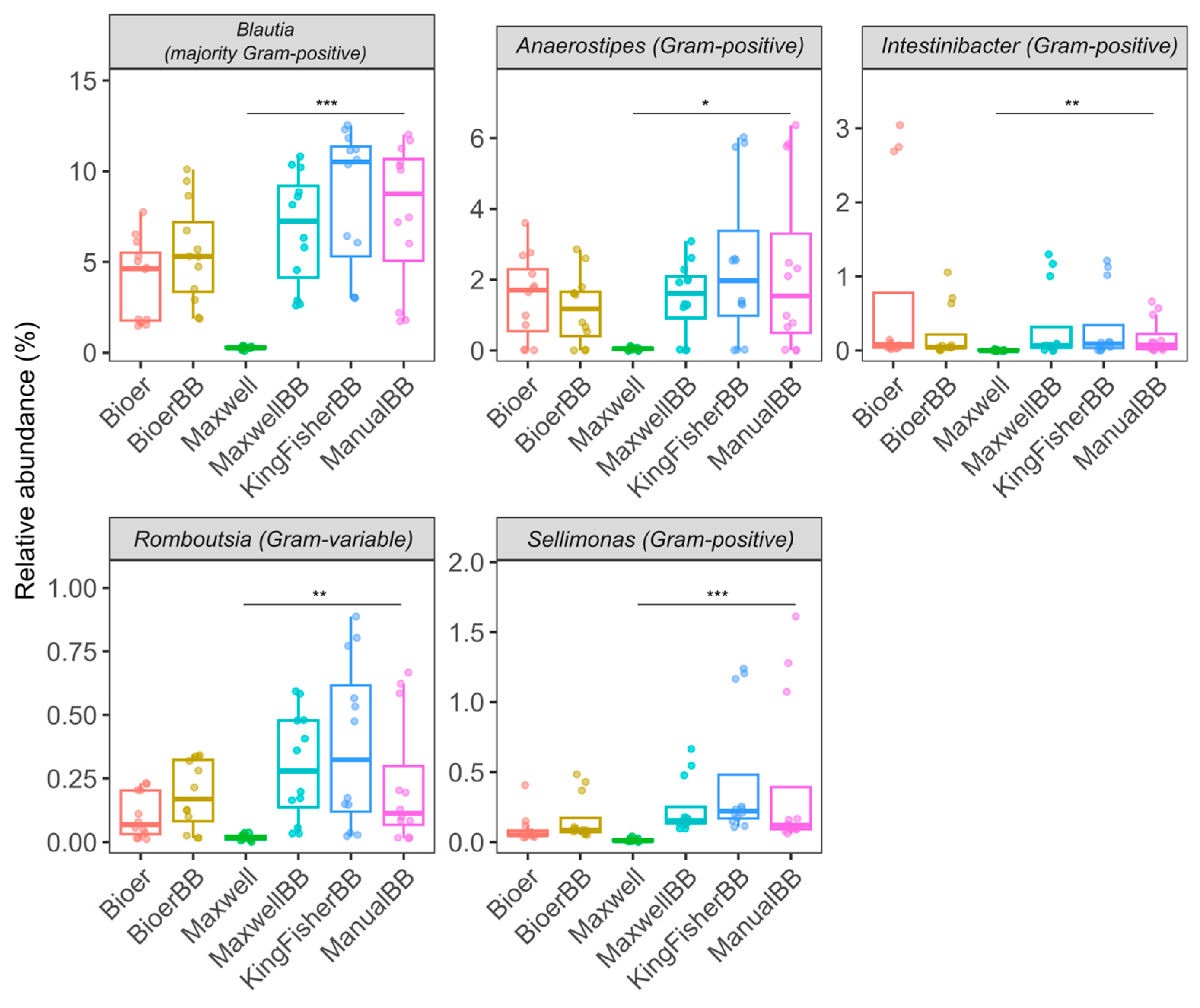
3.5. A Higher Relative Abundance of Gram-Positive Bacteria in Bead-Beaten Fecal Samples
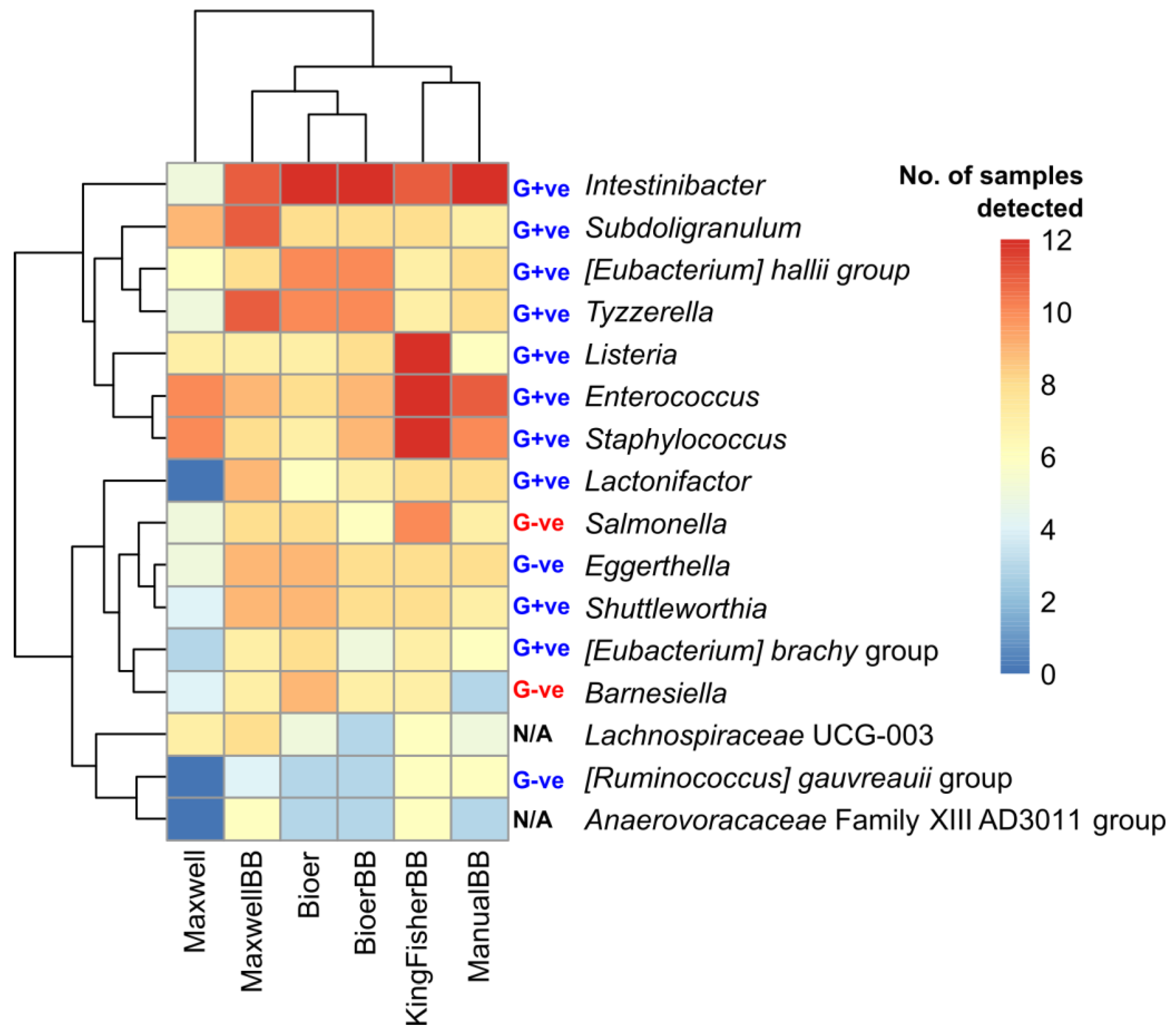
3.6. Heterogeneity in Prevalent Bacterial Genera Among Extraction Methods
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Bioer GenePure Pro Nucleic Acid Purification System (Catalog No. NPA-32P)
Appendix A.2. Promega Maxwell RSC DNA/RNA Extraction System (Catalog No: AS4500)
Appendix A.3. KingFisher Apex Purification Systems (ThermoFisher Scientific), (Catalog No: 5400910)
References
- Burckhardt, I. Laboratory Automation in Clinical Microbiology. Bioengineering 2018, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.; Davies, J.A. Automation in the Life Science Research Laboratory. Front. Bioeng. Biotechnol. 2020, 8, 571777. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gottardo, R. Comparability and reproducibility of biomedical data. Brief Bioinform. 2013, 14, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Wilke, W.W.; Jones, R.N.; Sutton, L.D. Automation of polymerase chain reaction tests. Reduction of human errors leading to contamination. Diagn Microbiol. Infect. Dis. 1995, 21, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Knepp, J.H.; Geahr, M.A.; Forman, M.S.; Valsamakis, A. Comparison of automated and manual nucleic acid extraction methods for detection of enterovirus RNA. J. Clin. Microbiol. 2003, 41, 3532–3536. [Google Scholar] [CrossRef]
- Sinke, L.; Cats, D.; Heijmans, B.T. Omixer: Multivariate and reproducible sample randomization to proactively counter batch effects in omics studies. Bioinformatics 2021, 37, 3051–3052. [Google Scholar] [CrossRef]
- Ling, W.; Lu, J.; Zhao, N.; Lulla, A.; Plantinga, A.M.; Fu, W.; Zhang, A.; Liu, H.; Song, H.; Li, Z.; et al. Batch effects removal for microbiome data via conditional quantile regression. Nat. Commun. 2022, 13, 5418. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef]
- Wang, Y.; Le Cao, K.A. PLSDA-batch: A multivariate framework to correct for batch effects in microbiome data. Brief Bioinform. 2023, 24, bbac622. [Google Scholar] [CrossRef]
- Sinha, R.; Abu-Ali, G.; Vogtmann, E.; Fodor, A.A.; Ren, B.; Amir, A.; Schwager, E.; Crabtree, J.; Ma, S.; Microbiome Quality Control Project, C.; et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 2017, 35, 1077–1086. [Google Scholar] [CrossRef]
- Parys-Proszek, A.; Branicki, W.; Wolańska-Nowak, P.; Kupiec, T. Application of BioRobot M48 to forensic DNA extraction. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 58–59. [Google Scholar] [CrossRef]
- Fiebelkorn, K.R.; Lee, B.G.; Hill, C.E.; Caliendo, A.M.; Nolte, F.S. Clinical evaluation of an automated nucleic acid isolation system. Clin. Chem. 2002, 48, 1613–1615. [Google Scholar] [CrossRef]
- Grisold, A.J.; Leitner, E.; Muhlbauer, G.; Marth, E.; Kessler, H.H. Detection of methicillin-resistant Staphylococcus aureus and simultaneous confirmation by automated nucleic acid extraction and real-time PCR. J. Clin. Microbiol. 2002, 40, 2392–2397. [Google Scholar] [CrossRef]
- Lim, M.Y.; Park, Y.S.; Kim, J.H.; Nam, Y.D. Evaluation of fecal DNA extraction protocols for human gut microbiome studies. BMC Microbiol. 2020, 20, 212. [Google Scholar] [CrossRef] [PubMed]
- Fiedorova, K.; Radvansky, M.; Nemcova, E.; Grombirikova, H.; Bosak, J.; Cernochova, M.; Lexa, M.; Smajs, D.; Freiberger, T. The Impact of DNA Extraction Methods on Stool Bacterial and Fungal Microbiota Community Recovery. Front. Microbiol. 2019, 10, 821. [Google Scholar] [CrossRef]
- Videnska, P.; Smerkova, K.; Zwinsova, B.; Popovici, V.; Micenkova, L.; Sedlar, K.; Budinska, E. Stool sampling and DNA isolation kits affect DNA quality and bacterial composition following 16S rRNA gene sequencing using MiSeq Illumina platform. Sci. Rep. 2019, 9, 13837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Brock, M.; Arana, C.; Dende, C.; van Oers, N.S.; Hooper, L.V.; Raj, P. Impact of Bead-Beating Intensity on the Genus- and Species-Level Characterization of the Gut Microbiome Using Amplicon and Complete 16S rRNA Gene Sequencing. Front. Cell Infect. Microbiol. 2021, 11, 678522. [Google Scholar] [CrossRef]
- Roopnarain, A.; Mukhuba, M.; Adeleke, R.; Moeletsi, M. Biases during DNA extraction affect bacterial and archaeal community profile of anaerobic digestion samples. 3 Biotech 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Lim, M.Y.; Song, E.J.; Kim, S.H.; Lee, J.; Nam, Y.D. Comparison of DNA extraction methods for human gut microbial community profiling. Syst. Appl. Microbiol. 2018, 41, 151–157. [Google Scholar] [CrossRef]
- Santiago, A.; Panda, S.; Mengels, G.; Martinez, X.; Azpiroz, F.; Dore, J.; Guarner, F.; Manichanh, C. Processing faecal samples: A step forward for standards in microbial community analysis. BMC Microbiol. 2014, 14, 112. [Google Scholar] [CrossRef]
- Fernandez-Pato, A.; Sinha, T.; Gacesa, R.; Andreu-Sanchez, S.; Gois, M.F.B.; Gelderloos-Arends, J.; Jansen, D.B.H.; Kruk, M.; Jaeger, M.; Joosten, L.A.B.; et al. Choice of DNA extraction method affects stool microbiome recovery and subsequent phenotypic association analyses. Sci. Rep. 2024, 14, 3911. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Oksanen, F.J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R Package Version 2.4-3. 2007. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 October 2024).
- Martinez Arbizu, P.R. PairwiseAdonis: Pairwise Multilevel Comparison Using Adonis. Package Version 0.4. 2020. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 1 October 2024).
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-scale Meta-omics Studies. PLoS Comput. Biol. 2011, 17, e1009442. [Google Scholar] [CrossRef]
- Reimer, L.C.; Carbasse, J.S.; Koblitz, J.; Ebeling, C.; Podstawka, A.; Overmann, J. BacDive in 2022: The knowledge base for standardized bacterial and archaeal data. Nucleic Acids Res. 2022, 7, D741–D746. [Google Scholar] [CrossRef]
- Starke, R.; Jehmlich, N.; Alfaro, T.; Dohnalkova, A.; Capek, P.; Bell, S.L.; Hofmockel, K.S. Incomplete cell disruption of resistant microbes. Sci. Rep. 2019, 9, 5618. [Google Scholar] [CrossRef]
- Wu, J.; Zhu, J.; Yin, H.; Liu, X.; An, M.; Pudlo, N.A.; Martens, E.C.; Chen, G.Y.; Lubman, D.M. Development of an Integrated Pipeline for Profiling Microbial Proteins from Mouse Fecal Samples by LC-MS/MS. J. Proteome Res. 2016, 15, 3635–3642. [Google Scholar] [CrossRef]
- Bisanz, J.E.; Upadhyay, V.; Turnbaugh, J.A.; Ly, K.; Turnbaugh, P.J. Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell Host Microbe 2019, 26, 265–272.e4. [Google Scholar] [CrossRef]
- Low, A.; Soh, M.; Miyake, S.; Aw, V.Z.J.; Feng, J.; Wong, A.; Seedorf, H. Longitudinal changes in diet cause repeatable and largely reversible shifts in gut microbial communities of laboratory mice and are observed across segments of the entire intestinal tract. Int. J. Mol. Sci. 2021, 22, 5981. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, C.; Wong, S.; Tighe, S.; Arumugam, A.; Liu, B.; Parker, C.W.; Wood, J.M.; Singh, N.K.; Skorupa, D.J.; Peyton, B.M.; et al. Validating an Automated Nucleic Acid Extraction Device for Omics in Space Using Whole Cell Microbial Reference Standards. Front. Microbiol. 2020, 11, 1909. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A relevant minority for the maintenance of gut homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- He, Z.; Tang, C.; Chen, X.; Liu, H.; Yang, G.; Xiao, Z.; Li, W.; Deng, Y.; Jin, L.; Chen, H.; et al. Based on magnetic beads to develop the kit for extraction of high-quality cell-free DNA from blood of breast cancer patients. Mater. Express 2019, 9, 956–961. [Google Scholar] [CrossRef]
- Marotz, C.; Sharma, A.; Humphrey, G.; Gottel, N.; Daum, C.; Gilbert, J.A.; Eloe-Fadrosh, E.; Knight, R. Triplicate PCR reactions for 16S rRNA gene amplicon sequencing are unnecessary. Biotechniques 2019, 67, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Engelbrektson, A.; Kunin, V.; Wrighton, K.C.; Zvenigorodsky, N.; Chen, F.; Ochman, H.; Hugenholtz, P. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010, 4, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, S.; Motooka, D.; Watanabe, S.; Kubo, R.; Jung, N.; Midorikawa, Y.; Shinozaki, N.O.; Sawai, Y.; Takeda, A.K.; Nakamura, S. Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1–V2 and V3–V4 primer sets. BMC Genom. 2021, 22, 527. [Google Scholar] [CrossRef]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Genepure Pro | Maxwell RSC 16 | King Fisher Apex | Manual Extraction |
|---|---|---|---|---|
| Manufacturer | Bioer | Promega | Applied Biosystem | N/A |
| Methods | Magnetic Bead-Based | Magnetic Bead-Based | Magnetic Bead-Based | Spin Columns |
| Format | 96 Wells Pre-Packed Plate | Individual Pre-Packed Cartridge | 96 wells Self-Filled Plate | Individual Self-Filled |
| Sample Volume | 300 µL | 300 µL | 300 µL | 300 µL |
| Throughput (samples/run) | 1–32 | 1–16 | 1–96 | Variable |
| Automation Level | Semi-automatic | Semi-automatic | Semi-automatic | Manual |
| Preparation Time (Based on 16 samples) | ~25 min | ~35 min | ~40 min | N/A |
| Processing Time (Based on 16 samples) | ~35 min | ~42 min | ~40 min | ~100 min |
| Sterilization | Built-in UV | Built-in UV | Built-in UV | N/A |
| Type of Sample | Stool, Body Fluid | Stool, Body Fluid | Stool, Body Fluid | Stool, Soil |
| Kits Used | MagaBio Fecal Pathogens DNA Purification Kit (Bioer Technology, Hangzhou, China) | Maxwell RSC Fecal Microbiome DNA kit (Promega Corporation, Madison, WI, USA) | MagMAX Microbiome Ultra Kit (ThermoFisher Scientific, Waltham, MA, USA) | FastDNA Spin Kit for Soil (MP-Biomedicals, Santa Ana, CA, USA) |
| Homogenizer (Bead-Beating) Needed | Yes/No | Yes/No | Yes | Yes |
| Estimated Elution volume | 50 µL | 50–100 µL | 50–200 µL | 50–100 µL |
| Total Raw Reads for Stool Samples | ||||
| Without Bead-Beating | 1,807,207 | 1,730,795 | N/A | N/A |
| With Bead-Beating | 1,482,643 | 1,753,841 | 1,223,111 | 1,274,852 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwa, W.T.; Sim, C.K.; Low, A.; Lee, J.W.J. A Comparison of Three Automated Nucleic Acid Extraction Systems for Human Stool Samples. Microorganisms 2024, 12, 2417. https://doi.org/10.3390/microorganisms12122417
Kwa WT, Sim CK, Low A, Lee JWJ. A Comparison of Three Automated Nucleic Acid Extraction Systems for Human Stool Samples. Microorganisms. 2024; 12(12):2417. https://doi.org/10.3390/microorganisms12122417
Chicago/Turabian StyleKwa, Wit Thun, Choon Kiat Sim, Adrian Low, and Jonathan Wei Jie Lee. 2024. "A Comparison of Three Automated Nucleic Acid Extraction Systems for Human Stool Samples" Microorganisms 12, no. 12: 2417. https://doi.org/10.3390/microorganisms12122417
APA StyleKwa, W. T., Sim, C. K., Low, A., & Lee, J. W. J. (2024). A Comparison of Three Automated Nucleic Acid Extraction Systems for Human Stool Samples. Microorganisms, 12(12), 2417. https://doi.org/10.3390/microorganisms12122417