
PhyloFunDB: A Pipeline to Create and Update Functional Gene Taxonomic Databases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
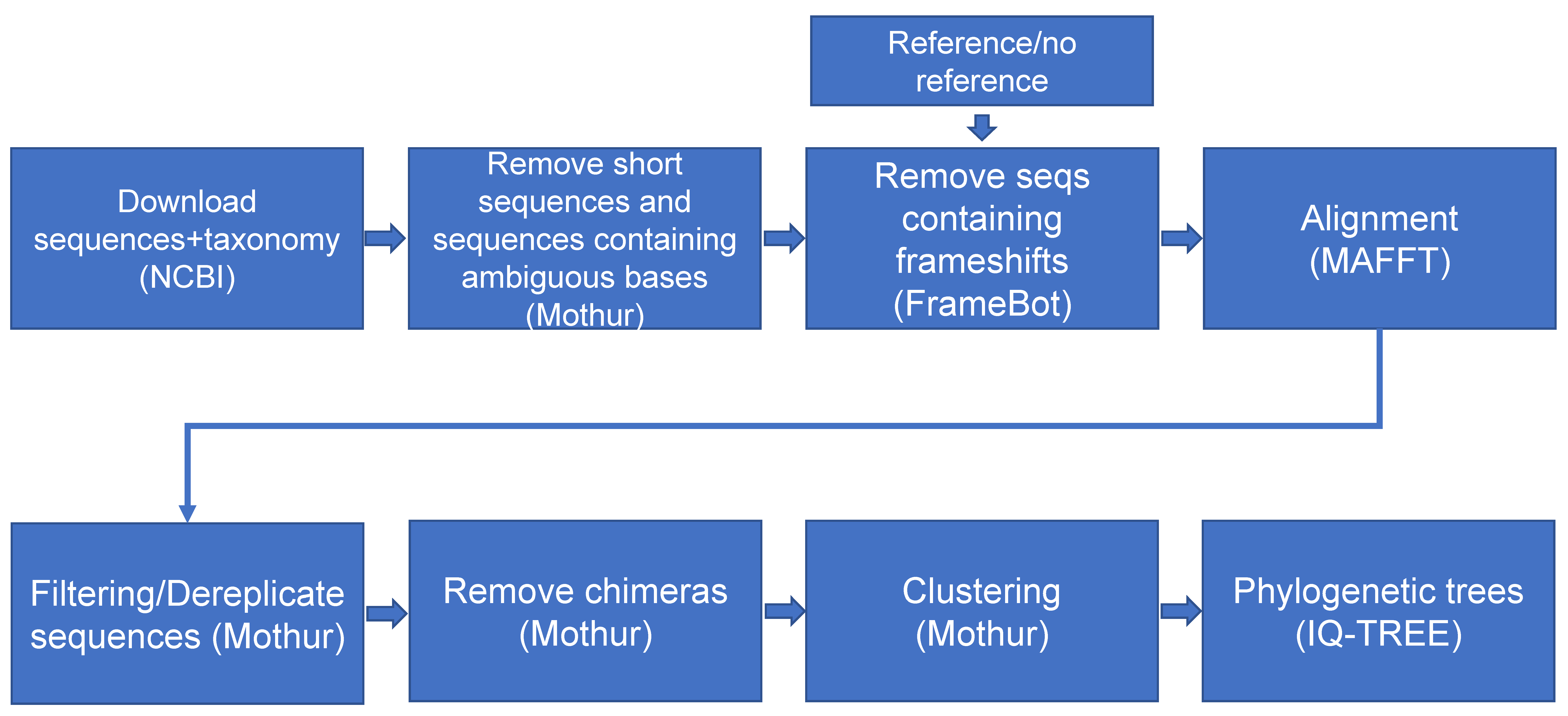
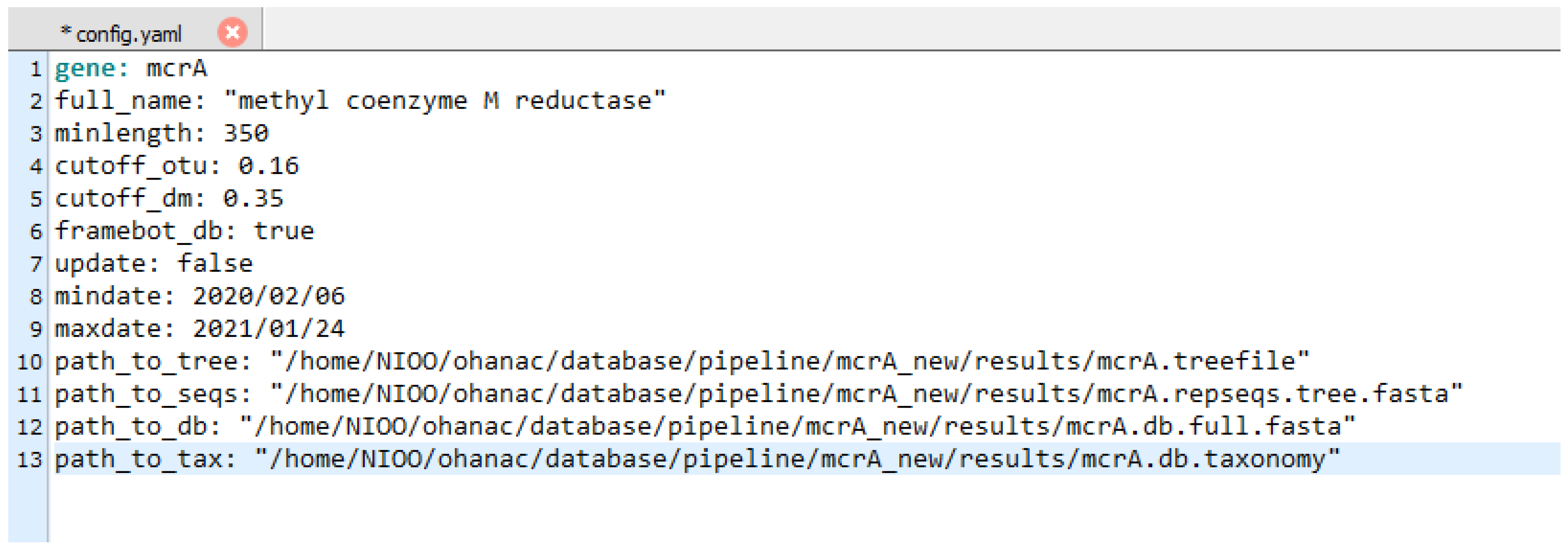
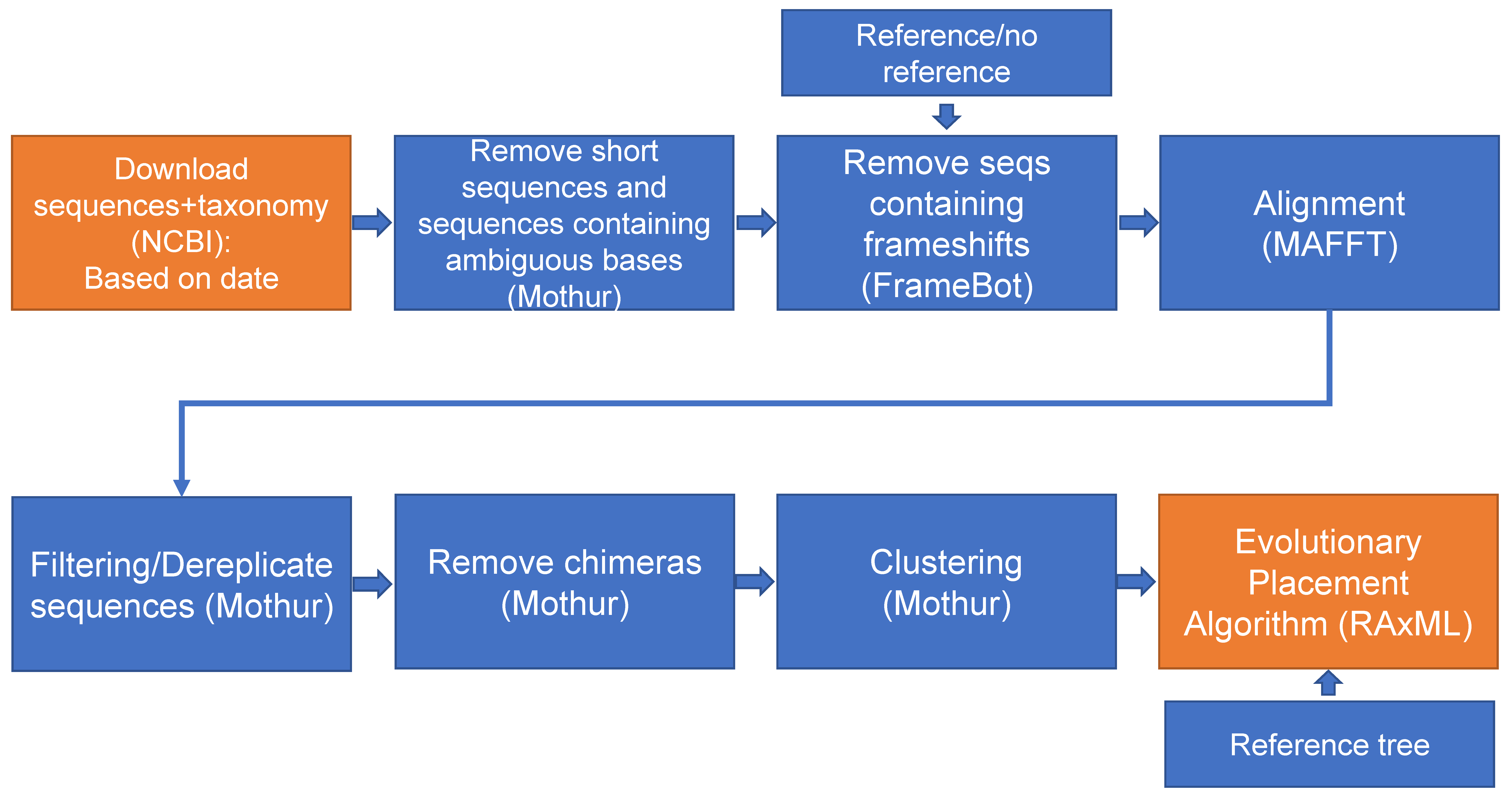
2. Materials and Methods
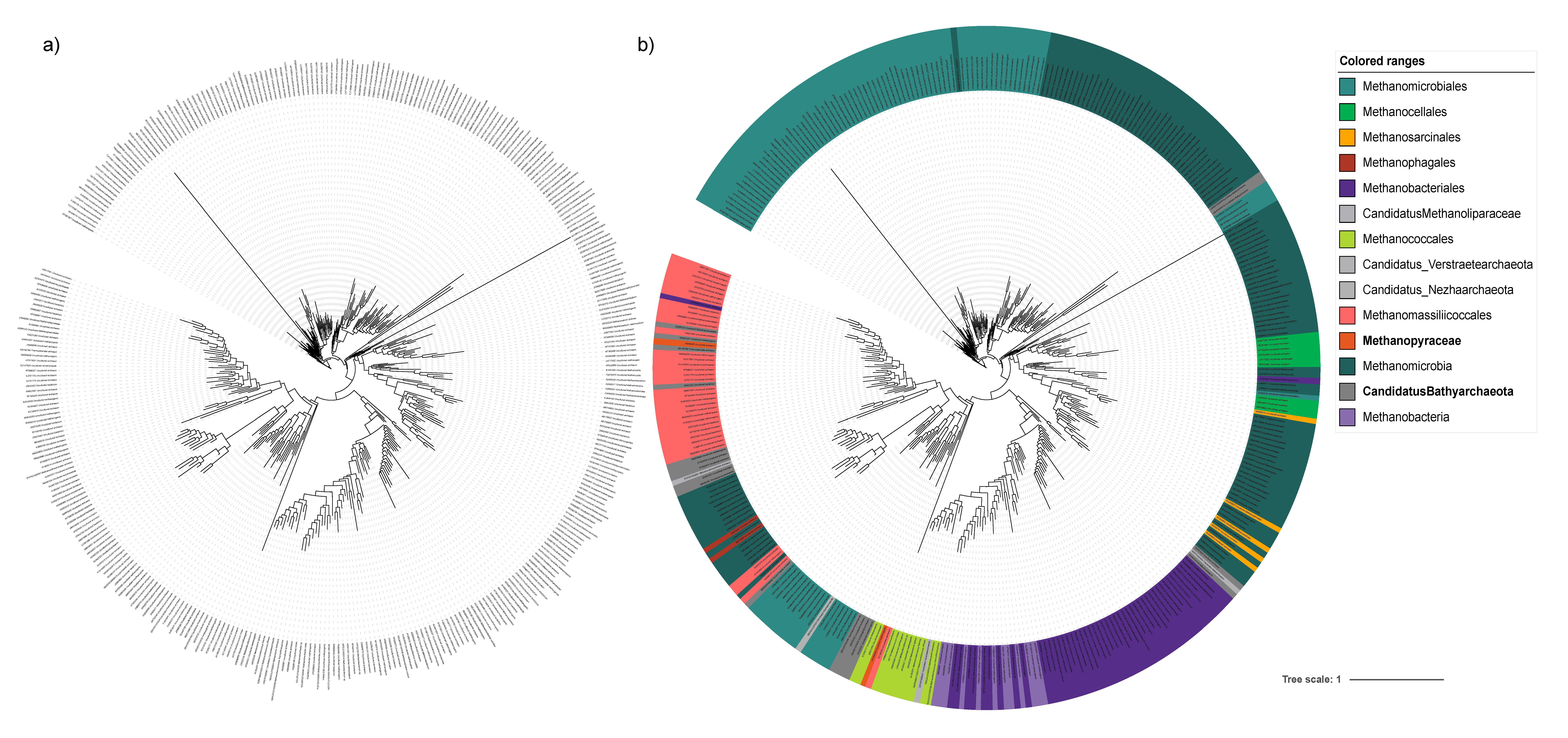
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tu, Q.; Lin, L.; Cheng, L.; Deng, Y.; He, Z. NCycDB: A Curated Integrative Database for Fast and Accurate Metagenomic Profiling of Nitrogen Cycling Genes. Bioinformatics 2019, 35, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Hallin, S.; Philippot, L.; Löffler, F.E.; Sanford, R.A.; Jones, C.M. Genomics and Ecology of Novel N2O-Reducing Microorganisms. Trends Microbiol. 2018, 26, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yang, S.; Horn, F.; Winkel, M.; Wagner, D.; Liebner, S. Global Biogeographic Analysis of Methanogenic Archaea Identifies Community-Shaping Environmental Factors of Natural Environments. Front. Microbiol. 2017, 8, 1339. [Google Scholar] [CrossRef] [PubMed]
- Knief, C. Diversity and Habitat Preferences of Cultivated and Uncultivated Aerobic Methanotrophic Bacteria Evaluated Based on PmoA as Molecular Marker. Front. Microbiol. 2015, 6, 1346. [Google Scholar] [CrossRef] [Green Version]
- Boyd, J.A.; Jungbluth, S.P.; Leu, A.O.; Evans, P.N.; Woodcroft, B.J.; Chadwick, G.L.; Orphan, V.J.; Amend, J.P.; Rappé, M.S.; Tyson, G.W. Divergent Methyl-Coenzyme M Reductase Genes in a Deep-Subseafloor Archaeoglobi. ISME J. 2019, 13, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Speth, D.R.; Orphan, V.J. Metabolic Marker Gene Mining Provides Insight in Global McrA Diversity and, Coupled with Targeted Genome Reconstruction, Sheds Further Light on Metabolic Potential of the Methanomassiliicoccales. PeerJ 2018, 6, e5614. [Google Scholar] [CrossRef] [Green Version]
- Conrad, R. Microbial Ecology of Methanogens and Methanotrophs. In Advances in Agronomy; Academic Press: Cambridge, MA, USA, 2007; Volume 96, pp. 1–63. [Google Scholar]
- Thauer, R.K.; Kaster, A.-K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic Archaea: Ecologically Relevant Differences in Energy Conservation. Nat. Rev. Microbiol. 2008, 6, 579–591. [Google Scholar] [CrossRef]
- Alves, R.J.E.; Minh, B.Q.; Urich, T.; von Haeseler, A.; Schleper, C. Unifying the Global Phylogeny and Environmental Distribution of Ammonia-Oxidising Archaea Based on AmoA Genes. Nat. Commun. 2018, 9, 1517. [Google Scholar] [CrossRef] [Green Version]
- Koster, J.; Rahmann, S. Snakemake—A Scalable Bioinformatics Workflow Engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E. A General Introduction to the E-Utilities; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Quensen, J.F.; Fish, J.A.; Lee, T.K.; Sun, Y.; Tiedje, J.M.; Cole, J.R. Ecological Patterns of NifH Genes in Four Terrestrial Climatic Zones Explored with Targeted Metagenomics Using FrameBot, a New Informatics Tool. mBio 2013, 4, e00592-13. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Liebner, S.; Alawi, M.; Ebenhöh, O.; Wagner, D. Taxonomic Database and Cut-off Value for Processing McrA Gene 454 Pyrosequencing Data by MOTHUR. J. Microbiol. Methods 2014, 103, 3–5. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Berger, S.A.; Krompass, D.; Stamatakis, A. Performance, Accuracy, and Web Server for Evolutionary Placement of Short Sequence Reads under Maximum Likelihood. Syst. Biol. 2011, 60, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.-G.; Zhang, X.-D.; Cao, M.; Liu, F.; Qian, Y.; Zhang, S.-W. Comparison of Methods for Picking the Operational Taxonomic Units From Amplicon Sequences. Front. Microbiol. 2021, 12, 644012. [Google Scholar] [CrossRef]
- Löytynoja, A. Phylogeny-Aware Alignment with PRANK. In Multiple Sequence Alignment Methods; Russell, D.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1079, pp. 155–170. ISBN 978-1-62703-645-0. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y. HMM-FRAME: Accurate Protein Domain Classification for Metagenomic Sequences Containing Frameshift Errors. BMC Bioinform. 2011, 12, 198. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Warnow, T. Large-Scale Multiple Sequence Alignment and Tree Estimation Using SATé. In Multiple Sequence Alignment Methods; Russell, D.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1079, pp. 219–244. ISBN 978-1-62703-645-0. [Google Scholar]
- Costa, O.Y.A.; De Hollander, M. Nioo-Knaw/PhyloFunDB: 1.0; Zenodo: Prévessin-Moëns, France, 2022. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, O.Y.A.; de Hollander, M.; Kuramae, E.E.; Bodelier, P.L.E. PhyloFunDB: A Pipeline to Create and Update Functional Gene Taxonomic Databases. Microorganisms 2022, 10, 1093. https://doi.org/10.3390/microorganisms10061093
Costa OYA, de Hollander M, Kuramae EE, Bodelier PLE. PhyloFunDB: A Pipeline to Create and Update Functional Gene Taxonomic Databases. Microorganisms. 2022; 10(6):1093. https://doi.org/10.3390/microorganisms10061093
Chicago/Turabian StyleCosta, Ohana Y. A., Mattias de Hollander, Eiko E. Kuramae, and Paul L. E. Bodelier. 2022. "PhyloFunDB: A Pipeline to Create and Update Functional Gene Taxonomic Databases" Microorganisms 10, no. 6: 1093. https://doi.org/10.3390/microorganisms10061093
APA StyleCosta, O. Y. A., de Hollander, M., Kuramae, E. E., & Bodelier, P. L. E. (2022). PhyloFunDB: A Pipeline to Create and Update Functional Gene Taxonomic Databases. Microorganisms, 10(6), 1093. https://doi.org/10.3390/microorganisms10061093