The Neuropathic Itch Caused by Pseudorabies Virus


Abstract
1. History of Aujeszky’s Disease
2. Case Reports of Neuropathic Itch Caused by PRV Infection in Non-Natural Hosts
3. Pseudorabies Virus (PRV)

{kind=link}
{kind=link}
{kind=link}
| Case n° | Year of Occurrence | Country | Species and Number of Confirmed Cases | Source of Contamination | Characteristic Clinical Symptoms | Gross Pathology | Histological Findings | Death after Onset of Clinical Symptoms | PRV Detection | Publication |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2004 | Poland | Farm animals: 7 cattle, 3 goats, 3 sheep, 2 cats and 1 dog | Not clear | -Local pruritus with violent licking, chewing and rubbing of various body parts -Fever -Excessive salivation | Not specified | Not specified | 48 h | Virus isolation and PCR on brain and internal organs samples | [24] |
| 2 | 2006 | Austria | 1 hunting dog | Contact with feral swine while hunting | -Severe pruritus on the lip with self-mutilation -Fever -Tachypnea -Seizures | -Purulent edema of the lip -Hypertrophied heart and hyperemia | -Non purulent encephalitis with intranuclear inclusion bodies -Multifocal lymphohistiocytic perivascular infiltrate in the medulla oblongata -Multifocal necrosis ganglia | 24 h | H&E staining, IHC, virus isolation and PCR on brain samples | [25,26] |
| 3 | 2007 | Belgium | 2 hunting dogs | Eaten offal from a wild boar | -Intense facial pruritus with self-mutilation -Convulsions -Hypersalivation | Not specified | Not specified | 24 h | Virus isolation and qPCR on brain samples | [27] |
| 4 | 2008 | Spain | 30 minks | Eaten swine viscera incorporated to food mixture | Not specified | Hemorrhages, ischemia and systemic vasculopathy | Mild purulent ganglioneuritis and encephalomyelitis | 48 h | IHC and PCR on oropharyngeal mucosa, TG, spinal cord and brain samples | [28] |
| 5 | 2008–2010 | Austria | 6 hunting dogs | Contact to the shot boars | -Intense pruritus on the neck and shoulders with self-mutilation -Hypersalivation -Coma | -Itch associated lesions in the head area -Purulent inflammation and edema of the subcutaneous tissues | Non purulent encephalitis of the brainstem with intranuclear inclusion bodies | 16–44 h | IHC and PCR on brain samples | [25,29] |
| 6 | 2011 | USA | 3 hunting dogs | Contact with feral swine while hunting | -Intense facial pruritus with self-mutilation -Fever -Dyspnea -Vomiting -Muscle stiffness | Extensive subendocardial hemorrhage | Neutrophilic trigeminal ganglioneuritis | 48 h | IHC and IF on TG samples | [30] |
| 7 | 2011–2013 | China | 13 farm and pet dogs | Possible contact with infected pigs or consumption of raw meat | -Pruritus -Tachypnea -Dyspnea | Systemic hemorrhage | -Non suppurative encephalitis with severe perivascular cuffing and glia cell proliferation | Not specified | Virus isolation and PCR on brain samples | [31] |
| 8 | 2012 | Germany | 1 hunting dog | Not specified | -Tremors -Trismus -Spasms of the musculature of the larynx and pharynx -Dyspnea -Vomiting | Not specified | Non suppurative encephalitis in the brainstem with perivascular cuffing of lymphocytes and macrophages | Not specified | IHC on TG and brain samples | [32] |
| 9 | 2012 | China | 860 sheep | Vaccinated with live-attenuated PRV-Bartha K16 strain | -Intense rubbing and licking with self-mutilation in the area where the vaccine was injected -Localized pruritus -Fever -Paralysis -Dyspnea | None | Not specified | 24 h | PCR and EM on brain samples | [33] |
| 10 | 2010–2014 | Italy | 11 hunting dogs | Direct contact with infected pigs or fed with raw meat | -Intense facial pruritus -Tremors -Dyspnea | Acute pulmonary alveolar emphysema and edema | Not specified | 24–48 h | IHC on brain samples | [34] |
| 11 | 2014 | Italy | 1 wild fox | Contact with infected domestic swine/feeding on infected rodents | -Head scratching -Motor coordination -Rolling in the snow | -Subcutaneous edema (head) -Multiple skin abrasions from scratching | Not specified | 48 h | Virus isolation on brain samples | [35] |
| 12 | 2014 | China | 379 minks | Captive minks fed raw pork livers | -Abdominal and facial pruritus -Claw and cage biting -Dyspnea- Vomiting | -Systemic hemorrhage -Splenic lesions -Petechia and ecchymoses in the epicardia | Not specified | 24–48 h | Virus isolation and IF on brain samples | [36] |
| 13 | 2014 | China | 3522 minks | Fed raw pork meat? | -Pneumonia like symptoms -Diarrhea -Lethargy | Not specified | Not specified | 48 h | Virus isolation and PCR on brain and internal organs samples | [37] |
| 14 | 2014 | China | 1200 captive foxes | Fed raw pork livers | -Initial fever -Intense pruritus -Frequent snarling and repeated lying down and rising -Dyspnea -Vomiting | Not specified | Not specified | 12–96 h | Virus isolation and PCR on brain samples | [38] |
| 15 | 2014 | USA | 10 hunting dogs | Contact with feral swine while hunting and consumption of pig offal | -Intense pruritus with self-mutilation -Erythema -Vomiting | Not specified | Moderate lymphoplasmatic encephalitis in the brainstem | 24 h | IHC, virus isolation and PCR on brain samples | [39] |
| 16 | 2017 | Spain | 1 Iberian Lynx | Eaten raw pig meat or offal? | Signs of scratching in the neck with self-mutilation | -Congested meninges -Multifocal erosions of the duodenum | -Meningoencephalitis with neutrophil and mononuclear cell infiltration -Gastrointestinal tract lesions | Found dead | PCR and IHC on brain, tonsil and intestinal samples | [40] |
| 17 | 2017 | China | 1 wolf | Fed pork or pig offal | -Intense pruritus -Paroxysmal convulsions -Quadriplegia -Dyspnea -Vomiting | -Hemorrhagic spots and necrosis in the liver -Hyperemia -Hemorrhages and edema in the meninges | Not specified | 6 h | PCR on brain, tonsil and lung samples | [41] |
| 18 | 2017 | Serbia | 1 domestic dog | Fed pig offal | -Pruritus in the head and neck -Ataxia | Focal pulmonary, gastric and renal hemorrhages | Not specified | 24 h | Virus isolation and PCR on brain and internal organ samples | [42] |
| 19 | 2017 | China | 1 human (female) | Eye contamination of pig sewage | -Fever -Headache -Visual impairment => Diagnosed with endophthalmitis => Under antiviral therapy (acyclovir) | Not applicable | Not applicable | The patient survived | NGS, Sanger sequencing and PCR on vitreous humor samples -PRV antibody test positive in CSF, 4 months after disease onset | [10] |
| 20 | 2018 | Argentina | 1 domestic dog | -Exposure to a serologically positive swine farm -Direct contact with pigs or fed raw pork meat | -Pruritus -Tremors -Trismus -Spasms of muscles of the larynx and pharynx -Dyspnea -Vomiting | Not specified | -Mononuclear cell infiltration in meninges -Mild diffuse gliosis -Neuronal satellitosis in gray matter | 24–48 h | Virus isolation and PCR on brain samples | [43] |
| 21 | 2018 | China | 9 cattle | Close contact to pig house | -Banging their head to the walls (scratching) -Ataxia -Gait and salivation | Leptomeningeal hyperaermia | -Non suppurative meningoencephalitis with mononuclear perivascular cuffing -Neuronal necrosis -Satellitosis | 24 h | PCR on brain samples | [44] |
| 22 | 2018 | China | 1 human (male) | Veterinarian, hands punctured by a knife used during autopsy of dead swine | -Fever -Headaches -Seizures -Coma within 3 days after the appearance of symptoms -Lumbar puncture indicated an opening pressure -CT brain imaging showed hypodensity in the bilateral basal ganglia => Diagnosed with viral encephalitis => Under antiviral therapy for 2 weeks (acyclovir) | Not applicable | Not applicable | The patient survived | PRV gB (serum and CSF) and gE (serum) antibodies detected at day 21 and 28 after disease onset | [45] |
| 23 | 2018 | China | 5 humans (4 males and 1 female) | -Contact with pigs at slaughterhouse -Hand injury at work | -Fever -Seizures -Respiratory failure -Visual impairment -Retinal necrosis -Brain MRI showed abnormal signs in the temporal lobes and insular cortex => All diagnosed with viral encephalitis => Under antiviral therapy (not specified) | Not applicable | Not applicable | All patients survived | NGS in CSF (2 to 37 PRV reads detected) | [46] |
| 24 | 2019 | China | 1 human (male) | Contact with pigs at work (sick pig handler) | -Fever -Convulsions -Pulmonary inflammation -Visual impairment -Retinal necrosis -Brain MRI showed hypo-intensity in the bilateral temporal lobe and hippocampus => Diagnosed with encephalitis complicated with bilateral retinal necrosis => Under antiviral therapy (acyclovir) | Not applicable | Not applicable | The patient survived | NGS in CSF (72 PRV reads detected) | [47] |
| 25 | 2020 | China | 1 human (male) | -Contact with pigs (pork vendor) -Hand cut | -Fever -Seizures -Coma within 1 day after the appearance of symptoms -Brain MRI showed abnormal signals in the bilateral frontal lobe, temporal lobe, insula lobe, basal ganglia and hippocampus -Inflammatory lesions in the bilateral hemisphere =>Diagnosed with viral encephalitis =>Under antiviral therapy for 2 weeks (acyclovir) | Not applicable | Not applicable | The patient survived | - NGS in CSF (74 reads detected) -PRV antibodies detected in plasma and CSF at 23 and 56 days after disease onset | [48] |
| 26 | 2018 | China | 4 humans (3 male and 1 female) | -Exposed to raw pork at work -1 injured during pork cutting | -Fever -Seizures -Coma within 1 to 4 days -Respiratory failure -Brain MRI showed abnormal signals mainly in the bilateral areas of the temporal lobe and bilateral basal ganglia -2/4=> Bilateral retinitis => All diagnosed with viral encephalitis => Under antiviral therapy for 2 weeks (acyclovir) | Not applicable | Not applicable | Three patients survived and 1 died | -NGS in CSF -PRV antibodies detected in available serum of 3 patients | [9] |
4. The Pathogenesis of PRV
4.1. Introduction
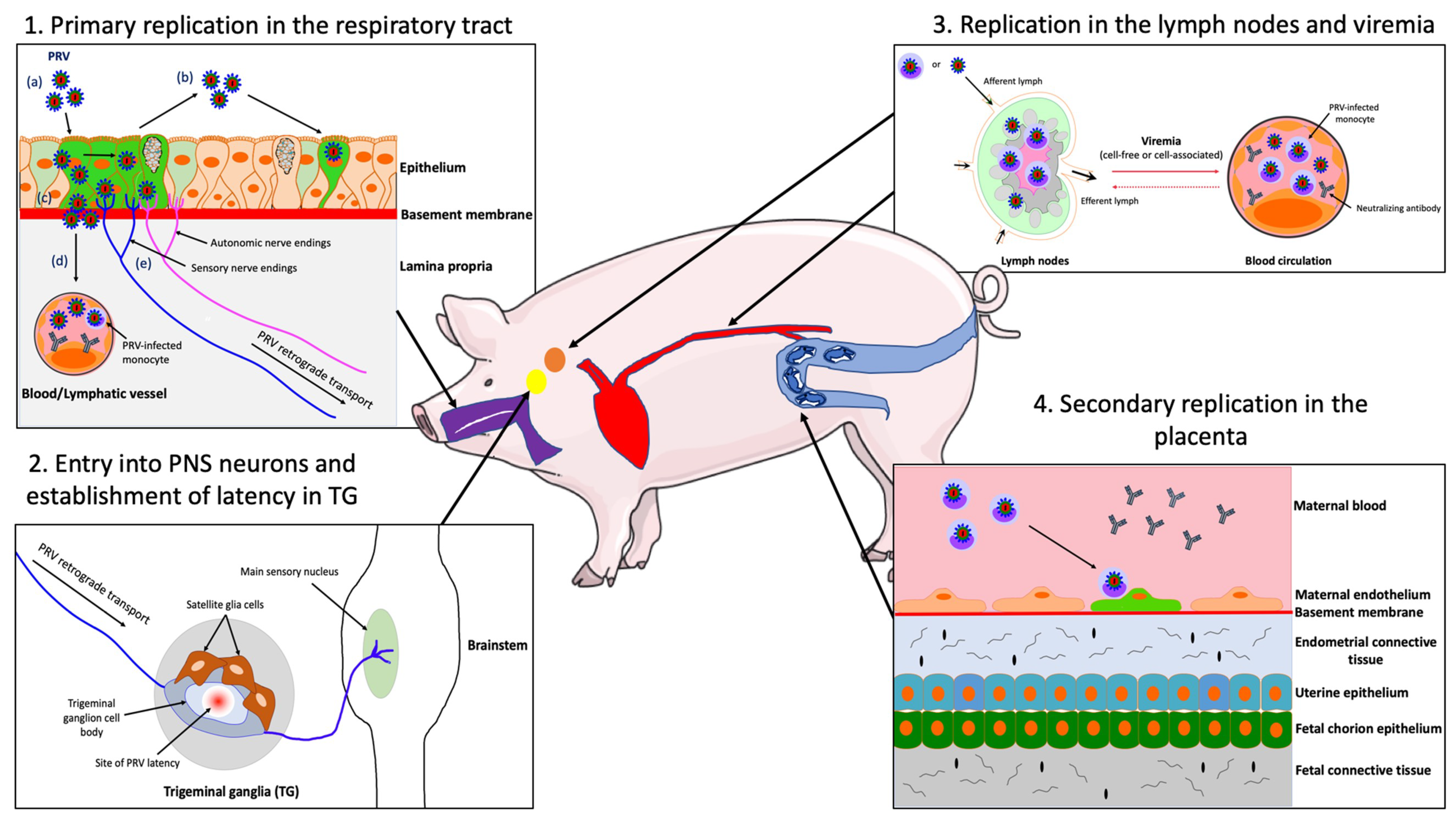
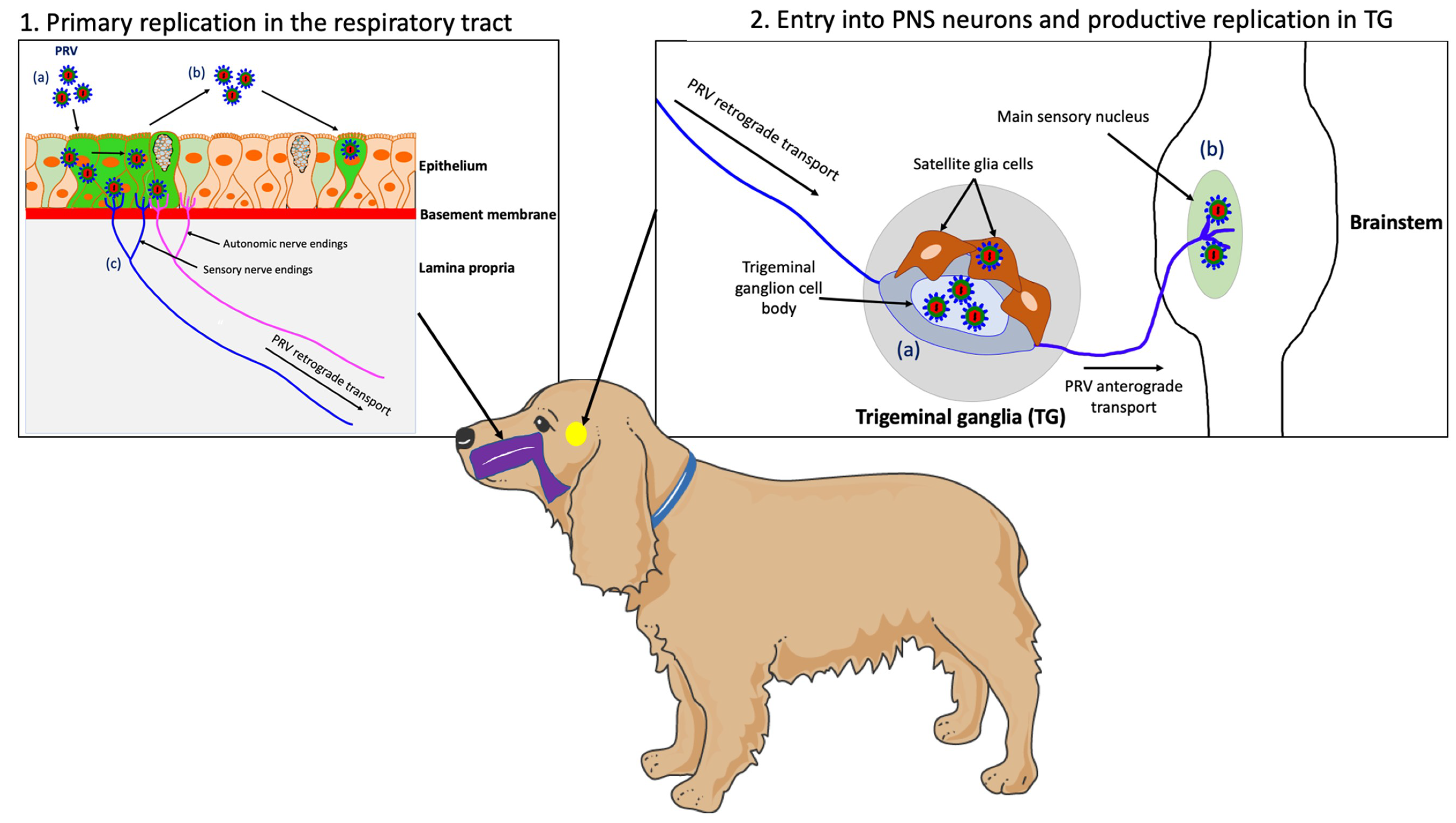
4.2. Primary Replication in the Upper Respiratory Tract (URT)
4.3. PRV Entry into the Peripheral Nervous Ssystem (PNS) Neurons and Spread to the Central Nervous System (CNS)
4.4. PRV Replication in the Draining Lymph Nodes and Viremia
4.5. Secondary Replication in the Swine Pregnant Vterus
4.6. PRV Infection in Suckling and Weaned Piglets
5. The Pathogenesis of PRV-Induced Neuropathic Itch
5.1. Clinical Classification of Itch
5.2. The Neuropathic Itch
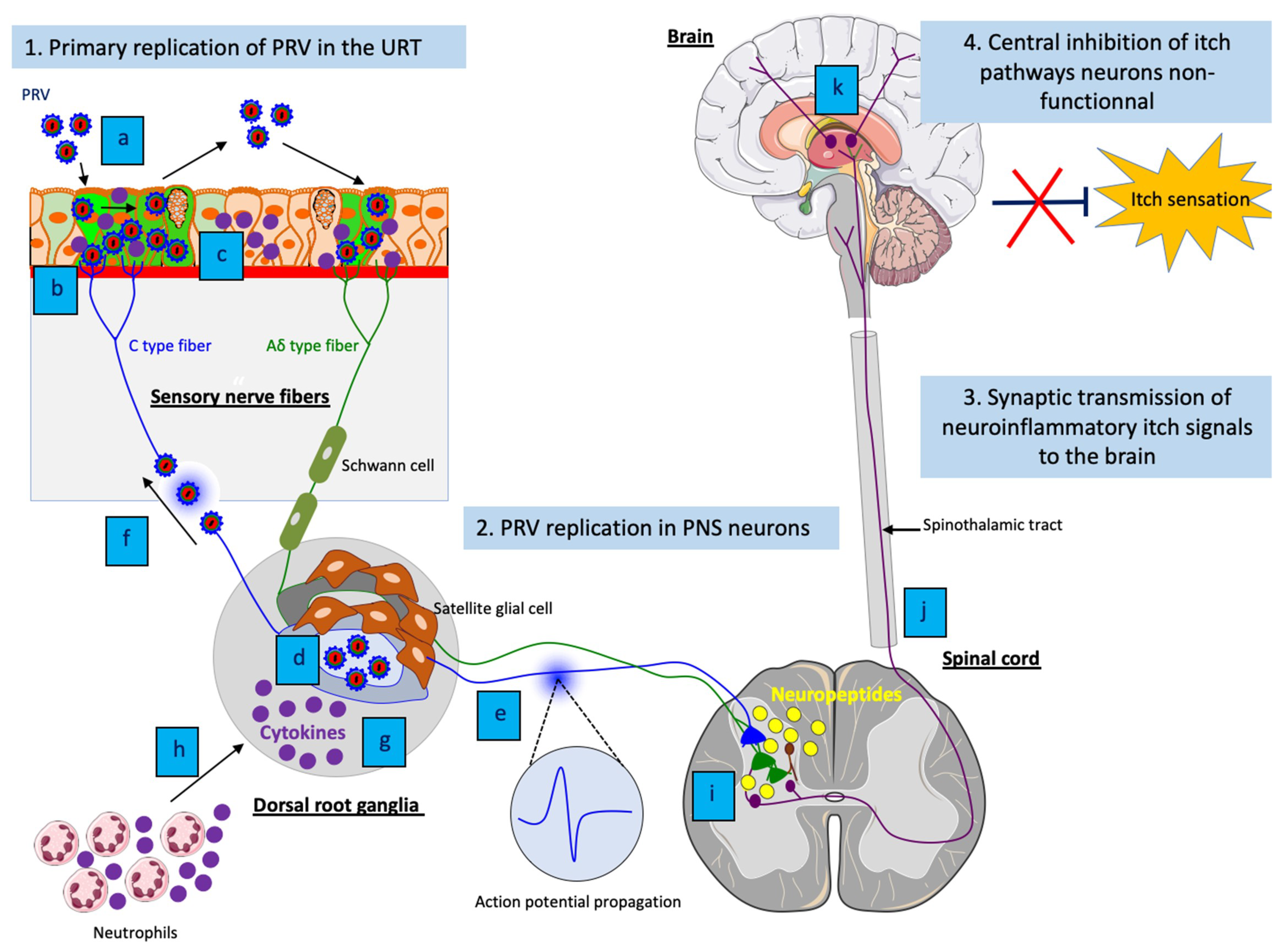
5.3. The Neuronal Mechanisms of PRV-Induced Neuropathic Itch
5.4. The Immune Mechanisms of PRV-Induced Neuropathic Itch
5.5. Why PRV-Infected Swine Do Not Itch
6. PRV Infection in Mice: A New Animal Model for VZV-Induced Peripheral Neuropathies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aujeszky, A. Uber eine neue Infektion krankheit bei Haustieren. Zbl Bakt Abt Orig 1902, 32, 353–357. [Google Scholar]
- Schmiedhoffer, J. Beiträge zur Pathologieder infektiösen Bulbär paralyse (Aujeszky-schen Krankheit). Z. Infekt. Krankh. Parasit. Hyg. Haustier 1910, 8, 383–405. [Google Scholar]
- Shope, R.E. An experimental study of ‘mad itch’ with especial reference to its relationship to pseudorabies. J. Exp. Med. 1931, 54, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Shope, R.E. Pseudorabies is a contagious disease in swine. Science 1934, 80, 102–103. [Google Scholar] [CrossRef] [PubMed]
- Sabin, A.B. Studies on the B virus. The immunological identity of a virus isolated from a human case of ascending myelitis associated with visceral necrosis. I. Br. J. Exp. Pathol. 1934, 15, 248–268. [Google Scholar]
- Sabin, A.B.A.W.; Wright, A.M. Acute ascending myelitis following monkey bite with isolation of a virus capable of reproducing disease. J. Exp. Med. 1934, 59, 115. [Google Scholar] [CrossRef]
- Sabin, A.B. Progression of different nasally installed viruses along different nervous pathways in the same host. Proc. Soc. Exp. Biol. Med. 1938, 38, 270–275. [Google Scholar] [CrossRef]
- Freuling, C.M.; Müller, T.F.; Mettenleiter, T.C. Vaccines against pseudorabies virus (PrV). Vet. Microbiol. 2017, 206, 3–9. [Google Scholar] [CrossRef]
- Zhao, W.L.; Wu, Y.H.; Li, H.F.; Li, S.Y.; Fan, S.Y.; Wu, H.L.; Li, Y.J.; Lu, Y.L.; Han, J. Clinical experience and next-generation sequencing analysis of encephalitis caused by pseudorabies virus. Zhonghua Yi Xue Za Zhi 2018, 98, 1152–1157. [Google Scholar]
- Ai, J.W.; Weng, S.S.; Cheng, Q.; Cui, P.; Li, Y.J.; Wu, H.L.; Zhu, Y.M.; Xu, B.; Zhang, W.H. Human Endophthalmitis Caused By Pseudorabies Virus Infection, China, 2017. Emerg. Infect. Dis. 2018, 24, 1087–1090. [Google Scholar] [CrossRef]
- Gu, J.; Hu, D.; Peng, T.; Wang, Y.; Ma, Z.; Liu, Z.; Meng, F.; Shang, Y.; Liu, S.; Xiao, Y. Epidemiological investigation of pseudorabies in Shandong Province from 2013 to 2016. Transbound Emerg Dis. 2018, 65, 890–898. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Xu, Q.; Wu, J.; Zhai, X.; Li, S.; Wang, J.; Ni, J.; Yuan, L.; Song, X.; et al. Investigation on pseudorabies prevalence in Chinese swine breeding farms in 2013–2016. Trop Anim. Health Prod. 2018, 50, 1279–1285. [Google Scholar] [CrossRef]
- Skinner, G.R.B.; Ahmad, A.; Davies, J.A. The infrequency of transmission of herpesviruses between humans and animals; postulation of an unrecognized protective host mechanism. Comp. Immunol. Microbiol. Infect. Dis. 2001, 24, 255–269. [Google Scholar] [CrossRef]
- Nauwynck, H.; Glorieux, S.; Favoreel, H.; Pensaert, M. Cell biological and molecular characteristics of pseudorabies virus infections in cell cultures and in pigs with emphasis on the respiratory tract. Vet. Res. 2007, 38, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C. Aujeszky’s disease (pseudorabies) virus: The virus and molecular pathogenesis—State of the art, June 1999. Vet. Res. 2000, 31, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G.E.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Roizmann, B.; Desrosiers, R.C.; Fleckenstein, B.; Lopez, C.; Minson, A.C.; Studdert, M.J. The family Herpesviridae: An update. The Herpesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch. Virol. 1992, 123, 425–449. [Google Scholar] [CrossRef]
- Ben-Porat, T.; Kaplan, A.S. Molecular Biology of Pseudorabies Virus. In The herpesviruses; Roizman, B., Ed.; Plenum Press: New York, NY, USA, 1985; Volume 3, pp. 105–173. [Google Scholar]
- Klupp, B.G.; Hengartner, C.J.; Mettenleiter, T.C.; Enquist, L.W. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 2004, 78, 424–440. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Herpesvirus assembly and egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef]
- Roizmann, B.F.D. The Replication of Herpesviruses. In Comprehensive Virology; Fraenkel-Conrat, H., RWagner, R., Eds.; Plenum Press: New York, NY, USA, 1974; pp. 229–403. [Google Scholar]
- Crabb, B.S.; Nagesha, H.S.; Studdert, M. Identification of equine herpesvirus 4 glycoprotein G: A type-specific, secreted glycoprotein. Virology 1992, 190, 143–154. [Google Scholar] [CrossRef]
- Salwa, A. A natural outbreak of Aujeszky’s disease in farm animals. Pol. J. Vet. Sci. 2004, 7, 261–266. [Google Scholar] [PubMed]
- Steinrigl, A.; Revilla-Fernandez, S.; Kolodziejek, J.; Wodak, E.; Bago, Z.; Nowotny, N.; Schmoll, F.; Kofer, J. Detection and molecular characterization of Suid herpesvirus type 1 in Austrian wild boar and hunting dogs. Vet. Microbiol. 2012, 157, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Thaller, D.; Bilek, A.; Revilla-Fernandez, S.; Bago, Z.; Schildorfer, H.; URL, A.; Weikel, J.; Weissenbock, H. Nachweis von Aujeszkyscher krankheit bei einem hund in Osterreich. Vet. Med. Austria. 2006, 93, 62–67. [Google Scholar]
- Cay, A.L.C. Isolation of anjeszky’s disease virus from two hunting dogs in Belgium after hunting wild boars. Vlaams Diergeneeskd. Tijdschr. 2009, 78, 194–195. [Google Scholar]
- Marcaccini, A.; Lopez Pena, M.; Quiroga, M.I.; Bermudez, R.; Nieto, J.M.; Aleman, N. Pseudorabies virus infection in mink: A host-specific pathogenesis. Vet. Immunol. Immunopathol. 2008, 124, 264–273. [Google Scholar] [CrossRef]
- Leschnik, M.; Gruber, A.; Kubber-Heiss, A.; Bago, Z.; Revilla-Fernandez, S.; Wodak, E.; Muller, E. Epidemiologische aspekte der Aujeskyschen krankheit in Osterrreich anhand von sechs aktuellen fallen beim hund. Wiener Tierarztliche Monatsschrift 2012, 99, 82–90. [Google Scholar]
- Cramer, S.D.; Campbell, G.A.; Njaa, B.L.; Morgan, S.E.; Smith, S.K., 2nd; McLin, W.R.t.; Brodersen, B.W.; Wise, A.G.; Scherba, G.; Langohr, I.M.; et al. Pseudorabies virus infection in Oklahoma hunting dogs. J. Vet. Diagn. Investig. 2011, 23, 915–923. [Google Scholar] [CrossRef]
- Zhang, L.; Zhong, C.; Wang, J.; Lu, Z.; Liu, L.; Yang, W.; Lyu, Y. Pathogenesis of natural and experimental Pseudorabies virus infections in dogs. J. Virol. 2015, 12, 44. [Google Scholar] [CrossRef]
- Schoniger, S.; Klose, K.; Werner, H.; Schwarz, B.A.; Muller, T.; Schoon, H.A. Nonsuppurative encephalitis in a dog. Vet. Pathol. 2012, 49, 731–734. [Google Scholar] [CrossRef]
- Kong, H.; Zhang, K.; Liu, Y.; Shang, Y.; Wu, B.; Liu, X. Attenuated live vaccine (Bartha-K16) caused pseudorabies (Aujeszky’s disease) in sheep. Vet. Res. Commun. 2013, 37, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Sozzi, E.; Grilli, G.; Gibelli, L.R.; Gelmetti, D.; Lelli, D.; Chiari, M.; Prati, P.; Alborali, G.L.; Boniotti, M.B.; et al. Detection and molecular analysis of Pseudorabies virus strains isolated from dogs and a wild boar in Italy. Vet. Microbiol. 2015, 177, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Dondo, A.; Cerutti, F.; Masoero, L.; Rosamilia, A.; Zoppi, S.; D’Errico, V.; Grattarola, C.; Acutis, P.L.; Peletto, S. Aujeszky’s Disease in Red Fox (Vulpes vulpes): Phylogenetic Analysis Unravels an Unexpected Epidemiologic Link. J. Wildl. Dis. 2014, 50, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, X.T.; Hu, B.; Deng, X.Y.; Zhang, L.; Lian, S.Z.; Zhang, H.L.; Lv, S.; Xue, X.H.; Lu, R.G.; et al. Outbreak of severe pseudorabies virus infection in pig-offal-fed farmed mink in Liaoning Province, China. Arch Virol. 2017, 162, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Du, Y.; Wu, J.Q.; Tian, F.L.; Yu, X.J.; Wang, J.B. Vaccine resistant pseudorabies virus causes mink infection in China. BMC Vet. Res. 2018, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.L.; Gao, S.M.; Liu, Y.; Zhang, S.F.; Hu, R.L. Pseudorabies in farmed foxes fed pig offal in Shandong province, China. Arch. Virol. 2016, 161, 445–448. [Google Scholar] [CrossRef]
- Pedersen, K.; Turnage, C.T.; Gaston, W.D.; Arruda, P.; Alls, S.A.; Gidlewski, T. Pseudorabies detected in hunting dogs in Alabama and Arkansas after close contact with feral swine (Sus scrofa). BMC Vet. Res. 2018, 14, 388. [Google Scholar] [CrossRef]
- Masot, A.J.; Gil, M.; Risco, D.; Jimenez, O.M.; Nunez, J.I.; Redondo, E. Pseudorabies virus infection (Aujeszky’s disease) in an Iberian lynx (Lynx pardinus) in Spain: A case report. BMC Vet. Res. 2017, 13, 6. [Google Scholar] [CrossRef]
- Lian, K.; Zhang, M.; Zhou, L.; Song, Y.; Wang, G.; Wang, S. First report of a pseudorabies-virus-infected wolf (Canis lupus) in China. Arch. Virol. 2019. [Google Scholar] [CrossRef]
- Lazic, G.; Lupulovic, D.; Topalski, B.; Bozić, B.; Lazic, S. Aujeszky’s disease in a dog—Case report. Arh. Vet. Med. 2017, 10, 61–69. [Google Scholar]
- Serena, M.S.; Metz, G.E.; Lozada, M.I.; Aspitia, C.G.; Nicolino, E.H.; Pidone, C.L.; Fossaroli, M.; Balsalobre, A.; Quiroga, M.A.; Echeverria, M.G. First isolation and molecular characterization of Suid herpesvirus type 1 from a domestic dog in Argentina. Open Vet. J. 2018, 8, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Kong, Z.; Liu, P.; Fu, Z.; Zhang, J.; Liu, M.; Shang, Y. Natural infection of a variant pseudorabies virus leads to bovine death in China. Transbound Emerg Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Han, H.; Wang, H.; Cui, Y.; Liu, H.; Ding, S.A. Case of Human Viral Encephalitis Caused by Pseudorabies Virus Infection in China. Front. Neurol. 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef]
- Wang, Y.; Nian, H.; Li, Z.; Wang, W.; Wang, X.; Cui, Y. Human encephalitis complicated with bilateral acute retinal necrosis associated with pseudorabies virus infection: A case report. Int. J. Infect. Dis. 2019, 89, 51–54. [Google Scholar] [CrossRef]
- Wang, D.; Tao, X.; Fei, M.; Chen, J.; Guo, W.; Li, P.; Wang, J. Human encephalitis caused by pseudorabies virus infection: A case report. J. Neurovirol. 2020. [Google Scholar] [CrossRef]
- Hahn, E.C.; Page, G.R.; Hahn, P.S.; Gillis, K.D.; Romero, C.; Annelli, J.A.; Gibbs, E.P. Mechanisms of transmission of Aujeszky’s disease virus originating from feral swine in the USA. Vet. Microbiol. 1997, 55, 123–130. [Google Scholar] [CrossRef]
- Romero, C.H.; Meade, P.N.; Shultz, J.E.; Chung, H.Y.; Gibbs, E.P.; Hahn, E.C.; Lollis, G. Venereal transmission of pseudorabies viruses indigenous to feral swine. J. Wildl. Dis. 2001, 37, 289–296. [Google Scholar] [CrossRef]
- Kluge, J.P.; Beran, G.W.; Hill, H.T.; Platt, K.B. Pseudorabies (Aujeszky’s Disease). In Diseases of swine, 8th ed.; Straw, B.E., D’Allaire, S., Mengeling, W.L., Taylor, D.J., Eds.; Iowa State University Press: Ames, IA, USA, 1999; pp. 233–246. [Google Scholar]
- Miry, C.P. Aujeszky’s Disease Virus Replication in Tonsils and Respiratory Tract of non-Immune and Immune Pigs. In Vaccination and Control of Aujeszky’s disease; van Oirschot, J.T., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1989; pp. 163–173. [Google Scholar]
- Wittmann, G. Aujeszky’s Disease (Pseudorabies) in Pigs. In Herpesvirus Diseases of Cattle, Horses and Pigs; Knipe, D.M., Howley, P.M., Eds.; Kluwer Academic Publishers: Boston, MA, USA, 1989; pp. 230–325. [Google Scholar]
- Masic, M.; Ercegan, M.; Petrovic, M. The significance of the tonsils in the pathogenesis and diagnosis of Aujeszyk’s disease in pigs. Zent. Vet. B 1965, 12, 398–405. [Google Scholar]
- Glorieux, S.; Van den Broeck, W.; van der Meulen, K.M.; Van Reeth, K.; Favoreel, H.W.; Nauwynck, H.J. In vitro culture of porcine respiratory nasal mucosa explants for studying the interaction of porcine viruses with the respiratory tract. J. Virol. Methods 2007, 142, 105–112. [Google Scholar] [CrossRef]
- Glorieux, S.; Favoreel, H.W.; Meesen, G.; de Vos, W.; Van den Broeck, W.; Nauwynck, H.J. Different replication characteristics of historical pseudorabies virus strains in porcine respiratory nasal mucosa explants. Vet. Microbiol. 2009, 136, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Kawashima, K.; Matsuura, S.; Uchimura, A.; Miura, Y. Pneumonia in pigs infected with pseudorabies virus and Haemophilus parasuis serovar 4. J. Comp. Pathol. 1994, 110, 329–339. [Google Scholar] [CrossRef]
- Maes, R.K.; Kanitz, C.L.; Gustafson, D.P. Shedding patterns in swine of virulent and attenuated pseudorabies virus. Am. J. Vet. Res. 1983, 44, 2083–2086. [Google Scholar] [PubMed]
- Babic, N.; Mettenleiter, T.C.; Ugolini, G.; Flamand, A.; Coulon, P. Propagation of pseudorabies virus in the nervous system of the mouse after intranasal inoculation. Virology 1994, 204, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Laval, K.; Van Cleemput, J.; Vernejoul, J.B.; Enquist, L.W. Alphaherpesvirus infection of mice primes PNS neurons to an inflammatory state regulated by TLR2 and type I IFN signaling. PLoS Pathog. 2019, 15, e1008087. [Google Scholar] [CrossRef]
- Brittle, E.E.; Reynolds, A.E.; Enquist, L.W. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J. Virol. 2004, 78, 12951–12963. [Google Scholar] [CrossRef]
- Field, H.J.; Hill, T.J. The pathogenesis of pseudorabies in mice following peripheral inoculation. J. Gen. Virol. 1974, 23, 145–157. [Google Scholar] [CrossRef]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral Microbiol. 2013, 5. [Google Scholar] [CrossRef]
- Tirabassi, R.S.; Townley, R.A.; Eldridge, M.G.; Enquist, L.W. Molecular mechanisms of neurotropic herpesvirus invasion and spread in the CNS. Neurosci. Biobehav. Rev. 1998, 22, 709–720. [Google Scholar] [CrossRef]
- Gutekunst, D.E.; Pirtle, E.C.; Miller, L.D.; Stewart, W.C. Isolation of pseudorabies virus from trigeminal ganglia of a latently infected sow. Am. J. Vet. Res. 1980, 41, 1315–1316. [Google Scholar]
- Wheeler, J.G.; Osorio, F.A. Investigation of sites of pseudorabies virus latency, using polymerase chain reaction. Am. J. Vet. Res. 1991, 52, 1799–1803. [Google Scholar] [PubMed]
- Van Oirschot, J.T.; Gielkens, A.L. In vivo and in vitro reactivation of latent pseudorabies virus in pigs born to vaccinated sows. Am. J. Vet. Res. 1984, 45, 567–571. [Google Scholar] [PubMed]
- Wigdahl, B.; Rong, B.L.; Kinney-Thomas, E. Varicella-zoster virus infection of human sensory neurons. Virology 1986, 152, 384–399. [Google Scholar] [CrossRef]
- Jones, C. Bovine Herpes Virus 1 (BHV-1) and Herpes Simplex Virus Type 1 (HSV-1) Promote Survival of Latently Infected Sensory Neurons, in Part by Inhibiting Apoptosis. J. Cell Death 2013, 6, 1–16. [Google Scholar] [CrossRef]
- Olson, G.R.; Miller, L.D. Studies on the pathogenesis of heart lesions in dogs infected with pseudorabies virus. Can. J. Vet. Res. 1986, 50, 245–250. [Google Scholar]
- Damann, N.; Klopfleisch, R.; Rothermel, M.; Doerner, J.F.; Mettenleiter, T.C.; Hatt, H.; Wetzel, C.H. Neuronal pathways of viral invasion in mice after intranasal inoculation of pseudorabies virus PrV-9112C2 expressing bovine herpesvirus 1 glycoprotein B. J. Neurovirol. 2006, 12, 60–64. [Google Scholar] [CrossRef]
- Lamote, J.A.S.; Glorieux, S.; Nauwynck, H.J.; Favoreel, H.W. The US3 Protein of Pseudorabies Virus Drives Viral Passage across the Basement Membrane in Porcine Respiratory Mucosa Explants. J. Virol. 2016, 90, 10945–10950. [Google Scholar] [CrossRef]
- Glorieux, S.; Favoreel, H.W.; Steukers, L.; Vandekerckhove, A.P.; Nauwynck, H.J. A trypsin-like serine protease is involved in pseudorabies virus invasion through the basement membrane barrier of porcine nasal respiratory mucosa. Vet. Res. 2011, 42, 58. [Google Scholar] [CrossRef]
- Jamrichova, O.; Skoda, R. Multiplication of pseudorabies virus in the inguinal lymph nodes of pigs. Acta Virol. 1968, 12, 555. [Google Scholar]
- Wittmann, G.; Ohlinger, V.; Rziha, H.J. Occurrence and reactivation of latent Aujeszky’s disease virus following challenge in previously vaccinated pigs. Arch. Virol. 1983, 75, 29–41. [Google Scholar] [CrossRef]
- Mulder, W.A.; Jacobs, L.; Priem, J.; Kok, G.L.; Wagenaar, F.; Kimman, T.G.; Pol, J.M. Glycoprotein gE-negative pseudorabies virus has a reduced capability to infect second- and third-order neurons of the olfactory and trigeminal routes in the porcine central nervous system. J. Gen. Virol. 1994, 75, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Rajcani, J.; Blaskovic, D. Studies on the pathogenesis of Aujeszky’s disease virus. III. The distribution of virulent virus in piglets after intranasal infection. Acta Virol. 1969, 13, 407–714. [Google Scholar] [PubMed]
- Nauwynck, H.J.; Pensaert, M.B. Interactions of Aujeszky’s disease virus and porcine blood mononuclear cells in vivo and in vitro. Acta Vet. Hung. 1994, 42, 301–308. [Google Scholar] [PubMed]
- Nawynck, H.J.; Pensaert, M.B. Cell-free and cell-associated viremia in pigs after oronasal infection with Aujeszky’s disease virus. Vet. Microbiol. 1995, 43, 307–314. [Google Scholar] [CrossRef]
- Mc Ferran, I.B.; Dow, C. Virus studies on experimental Aujeszky’s disease in calves. J. Comp. Pathol. 1964, 74, 173–179. [Google Scholar] [CrossRef]
- Laval, K.; Vernejoul, J.B.; Van Cleemput, J.; Koyuncu, O.O.; Enquist, L.W. Virulent Pseudorabies Virus Infection Induces a Specific and Lethal Systemic Inflammatory Response in Mice. J. Virol. 2018, 92, e01614-18. [Google Scholar] [CrossRef]
- Nauwynck, H.J.; Pensaert, M.B. Abortion induced by cell-associated pseudorabies virus in vaccinated sows. Am. J. Vet. Res. 1992, 53, 489–493. [Google Scholar]
- Kluge, J.P.; Mare, C.J. Swine pseudorabies: Abortion, clinical disease, and lesions in pregnant gilts infected with pseudorabies virus (Aujeszky’s disease). Am. J. Vet. Res. 1974, 35, 991–995. [Google Scholar]
- Hsu, F.S.; Chu, R.M.; Lee, R.C.; Chu, S.H. Placental lesions caused by pseudorabies virus in pregnant sows. J. Am. Vet. Med. Assoc. 1980, 177, 636–641. [Google Scholar]
- Van de Walle, G.R.; Favoreel, H.W.; Nauwynck, H.J.; Mettenleiter, T.C.; Pensaert, M.B. Transmission of pseudorabies virus from immune-masked blood monocytes to endothelial cells. J. Gen. Virol. 2003, 84, 629–637. [Google Scholar] [CrossRef]
- Ka, H.; Seo, H.; Choi, Y.; Yoo, I.; Han, J. Endometrial response to conceptus-derived estrogen and interleukin-1β at the time of implantation in pigs. J. Anim. Sci. Biotechnol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Velez, C.; Barbeito, C.; Koncurat, M. alphavbeta3 Integrin and fibronectin expressions and their relation to estrogen and progesterone during placentation in swine. Biotech. Histochem. 2018, 93, 15–24. [Google Scholar] [PubMed]
- Bidarimath, M.; Tayade, C. Pregnancy and spontaneous fetal loss: A pig perspective. Mol. Reprod. Dev. 2017, 84, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Dieuzy, I.; Vannier, P.; Jestin, A. Effects of experimental pseudorabies virus infection on vaccinated pregnant sows. Ann. Rech. Vet. 1987, 18, 233–240. [Google Scholar]
- Iglesias, J.G.; Harkness, J.W. Studies of transplacental and perinatal infection with two clones of a single Aujeszky’s disease (pseudorabies) virus isolate. Vet. Microbiol. 1988, 16, 243–254. [Google Scholar] [CrossRef]
- Ceriatti, F.S.; Sabini, L.I.; Bettera, S.G.; Zanon, S.M.; Ramos, B.A. Experimental infection of pregnant gilts with Aujeszky’s disease virus strain RC/79. Rev. Argent Microbiol. 1992, 24, 102–112. [Google Scholar]
- Ezura, K.; Usami, Y.; Tajima, K.; Komaniwa, H.; Nagai, S.; Narita, M.; Kawashima, K. Gastrointestinal and skin lesions in piglets naturally infected with pseudorabies virus. J. Vet. Diagn. Investig. 1995, 7, 451–455. [Google Scholar] [CrossRef]
- Ständer, S.; Weisshaar, E.; Mettang, T.; Szepietowski, J.C.; Carstens, E.; Ikoma, A.; Bergasa, N.V.; Gieler, U.; Misery, L.; Wallengren, J.; et al. Clinical classification of itch: A position paper of the International Forum for the Study of Itch. Acta Derm. Venereol. 2007, 87, 291–294. [Google Scholar] [CrossRef]
- Ikoma, A.; Steinhoff, M.; Ständer, S.; Yosipovitch, G.; Schmelz, M. The neurobiology of itch. Nat. Rev. Neurosci. 2006, 7, 535–547. [Google Scholar] [CrossRef]
- Grundmann, S.; Stander, S. Chronic pruritus: Clinics and treatment. Ann. Dermatol. 2011, 23, 1–11. [Google Scholar] [CrossRef]
- Green, D.; Dong, X. The cell biology of acute itch. J. Cell. Biol. 2016, 213, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yosipovitch, G.; Fleischer, A. Itch associated with skin disease: Advances in pathophysiology and emerging therapies. Am. J. Clin. Dermatol. 2003, 4, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Galatian, A.; Stearns, G.; Grau, R. Pruritus in connective tissue and other common systemic disease states. Cutis 2009, 84, 207–214. [Google Scholar] [PubMed]
- Binder, A.; Koroschetz, J.; Baron, R. Disease mechanisms in neuropathic itch. Nat. Clin. Pract. Neurol. 2008, 4, 329–337. [Google Scholar] [CrossRef]
- Yosipovitch, G.; Samuel, L.S. Neuropathic and psychogenic itch. Dermatol. Ther. 2008, 21, 32–41. [Google Scholar] [CrossRef]
- Oaklander, A.L. Neuropathic itch. Semin. Cutan. Med. Surg. 2011, 30, 87–92. [Google Scholar] [CrossRef]
- Ringkamp, M.; Meyer, R. Itch: Mechanisms and treatment; Carstens, E., Akiyama, T., Eds.; Frontiers in Neuroscience Pruriceptors; CRC Press/Taylor & Francis(c), LLC: Boca Raton, FL, USA, 2014. [Google Scholar]
- Robbins, B.A.; Schmieder, G.J. Brachioradial Pruritus. In StatPearls; StatPearls Publishing StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Mittal, A.; Srivastava, A.; Balai, M.; Khare, A.K. A study of postherpetic pruritus. Indian Dermatol. Online J. 2016, 7, 343–344. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yabuki, S.; Hayabara, T.; Otsuki, S. Paroxysmal itching in multiple sclerosis: A report of three cases. J. Neurol. Neurosurg. Psychiatry 1981, 44, 19–22. [Google Scholar] [CrossRef]
- Koeppel, M.C.; Bramont, C.; Ceccaldi, M.; Habib, M.; Sayag, J. Paroxysmal pruritus and multiple sclerosis. Br. J. Dermatol. 1993, 129, 597–598. [Google Scholar] [CrossRef]
- King, C.A.; Huff, F.J.; Jorizzo, J.L. Unilateral neurogenic pruritus: Paroxysmal itching associated with central nervous system lesions. Ann. Intern. Med. 1982, 97, 222–223. [Google Scholar] [CrossRef]
- Steinhoff, M.; Schmelz, M.; Szabó, I.L.; Oaklander, A.L. Clinical presentation, management, and pathophysiology of neuropathic itch. Lancet Neurol. 2018, 17, 709–720. [Google Scholar] [CrossRef]
- Davidson, S.; Zhang, X.; Khasabov, S.G.; Simone, D.A.; Giesler, G.J., Jr. Relief of itch by scratching: State-dependent inhibition of primate spinothalamic tract neurons. Nat. Neurosci. 2009, 12, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Yosipovitch, G.; Ishiuji, Y.; Patel, T.S.; Hicks, M.I.; Oshiro, Y.; Kraft, R.A.; Winnicki, E.; Coghill, R.C. The Brain Processing of Scratching. J. Investig. Dermatol. 2019, 128, 1806–1811. [Google Scholar] [CrossRef] [PubMed]
- Dempsher, J.; Larrabee, M.G.; Bang, F.B.; Bodian, D. Physiological changes in sympathetic ganglia infected with pseudorabies virus. Am. J. Physiol. 1955, 182, 203–216. [Google Scholar] [CrossRef]
- Tokumaru, T. Pseudorabies virus—Induced neural hyperreactivity following occular and skin infections in the rat. Res. Commun. Chem. Pathol. Pharmacol. 1975, 10, 533–542. [Google Scholar]
- Dolivo, M.; Beretta, E.; Bonifas, V.; Foroglou, C. Ultrastructure and function in sympathetic ganglia isolated from rats infected with pseudorabies virus. Brain Res. 1978, 140, 111–123. [Google Scholar] [CrossRef]
- Liao, G.S.; Maillard, M.; Kiraly, M. Ion channels involved in the presynaptic hyperexcitability induced by herpes virus suid in rat superior cervical ganglion. Neuroscience 1991, 41, 797–807. [Google Scholar] [CrossRef]
- McCarthy, K.M.; Tank, D.W.; Enquist, L.W. Pseudorabies virus infection alters neuronal activity and connectivity in vitro. PLoS Pathog. 2009, 5, e1000640. [Google Scholar] [CrossRef]
- Favoreel, H.W.; Van Minnebruggen, G.; Nauwynck, H.J.; Enquist, L.W.; Pensaert, M.B. A tyrosine-based motif in the cytoplasmic tail of pseudorabies virus glycoprotein B is important for both antibody-induced internalization of viral glycoproteins and efficient cell-to-cell spread. J. Virol. 2002, 76, 6845–6851. [Google Scholar] [CrossRef]
- Granstedt, A.E.; Bosse, J.B.; Thiberge, S.Y.; Enquist, L.W. In vivo imaging of alphaherpesvirus infection reveals synchronized activity dependent on axonal sorting of viral proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3516–E3525. [Google Scholar] [CrossRef]
- Brideau, A.D.; Card, J.P.; Enquist, L.W. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 2000, 74, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Husak, P.J.; Kuo, T.; Enquist, L.W. Pseudorabies virus membrane proteins gI and gE facilitate anterograde spread of infection in projection-specific neurons in the rat. J. Virol. 2000, 74, 10975–10983. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, M.; Card, J.P.; Tirabassi, R.S.; Miselis, R.R.; Enquist, L.W. Retrograde, transneuronal spread of pseudorabies virus in defined neuronal circuitry of the rat brain is facilitated by gE mutations that reduce virulence. J. Virol. 1999, 73, 4350–4359. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, M. Itch Processing in the Skin. Front. Med. 2019, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Ossipov, M.H.; Johnson, K.W. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 2017, 158, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Ge, T.T.; Fan, J.; Yang, W.; Cui, R.J. The role of substance P in epilepsy and seizure disorders. Oncotarget 2017, 8, 78225–78233. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.H.; Gilden, D.H.; Cohrs, R.J.; Mahalingam, R.; Nagel, M.A. Varicella zoster virus infection: Clinical features, molecular pathogenesis of disease, and latency. Neurol. Clin. 2008, 26, 675–697. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Yang, P.; Wen, H.; Ou, S.; Cui, J.; Fan, D. IL-6 promotes regeneration and functional recovery after cortical spinal tract injury by reactivating intrinsic growth program of neurons and enhancing synapse formation. Exp. Neurol. 2012, 236, 19–27. [Google Scholar] [CrossRef]
- Yong, K.L. Granulocyte colony-stimulating factor (G-CSF) increases neutrophil migration across vascular endothelium independent of an effect on adhesion: Comparison with granulocyte-macrophage colony-stimulating factor (GM-CSF). Br. J. Haematol. 1996, 94, 40–47. [Google Scholar] [CrossRef]
- Shaw, S.K.; Owolabi, S.A.; Bagley, J.; Morin, N.; Cheng, E.; LeBlanc, B.W.; Kim, M.; Harty, P.; Waxman, S.G.; Saab, C.Y. Activated polymorphonuclear cells promote injury and excitability of dorsal root ganglia neurons. Exp. Neurol. 2008, 210, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.J.; Lee, S.Y.; Choi, S.I.; Kim, B.K.; Lee, E.J.; In, K.H.; Lee, M.G. Toll-like receptor expression in pulmonary sensory neurons in the bleomycin-induced fibrosis model. PLoS ONE 2018, 13, e0193117. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Garssen, J.; Kraneveld, A.D. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol. Neuroinflamm. 2016, 3, 27–37. [Google Scholar] [CrossRef]
- Kim, D.; Kim, M.A.; Cho, I.H.; Kim, M.S.; Lee, S.; Jo, E.K.; Choi, S.Y.; Park, K.; Kim, J.S.; Akira, S.; et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem. 2007, 282, 14975–14983. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; Song, R.; Greco, T.M.; Cristea, I.M.; Enquist, L.W. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. MBio 2015, 6, e00276-15. [Google Scholar] [CrossRef] [PubMed]
- De Regge, N.; Van Opdenbosch, N.; Nauwynck, H.J.; Efstathiou, S.; Favoreel, H.W. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS ONE 2010, 5, e13076. [Google Scholar] [CrossRef]
- Pol, J.M.; Broekhuysen-Davies, J.M.; Wagenaar, F.; La Bonnardiere, C. The influence of porcine recombinant interferon-alpha 1 on pseudorabies virus infection of porcine nasal mucosa in vitro. J. Gen. Virol. 1991, 72, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; De Regge, N.; Van Poucke, M.; Peelman, L.; Favoreel, H.W. Effects of interferon on immediate-early mRNA and protein levels in sensory neuronal cells infected with herpes simplex virus type 1 or pseudorabies virus. Vet. Microbiol. 2011, 152, 401–406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, R.; Koyuncu, O.O.; Greco, T.M.; Diner, B.A.; Cristea, I.M.; Enquist, L.W. Two Modes of the Axonal Interferon Response Limit Alphaherpesvirus Neuroinvasion. MBio 2016, 7, e02145-15. [Google Scholar] [CrossRef]
- Lamote, J.A.S.; Kestens, M.; Van Waesberghe, C.; Delva, J.; De Pelsmaeker, S.; Devriendt, B.; Favoreel, H.W. The Pseudorabies Virus Glycoprotein gE/gI Complex Suppresses Type I Interferon Production by Plasmacytoid Dendritic Cells. J. Virol. 2017, 91, e02276-16. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.F.; Ming, S.L.; Li, G.L.; Zeng, L.; Wang, M.D.; Su, B.Q.; Wang, Q.; Yang, G.Y.; Chu, B.B. Porcine IFITM1 is a host restriction factor that inhibits pseudorabies virus infection. Int. J. Biol. Macromol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, S.; Yang, X.; Wang, X.; Li, Y.; Wang, C.; Chen, L.; Chang, H. Porcine ISG15 modulates the antiviral response during pseudorabies virus replication. Gene 2018, 679, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Gnann, J.W., Jr. Varicella-zoster virus: Atypical presentations and unusual complications. J. Infect. Dis. 2002, 186, S91–S98. [Google Scholar] [CrossRef] [PubMed]
- Kost, R.G.; Straus, S.E. Postherpetic neuralgia—Pathogenesis, treatment, and prevention. N. Engl. J. Med. 1996, 335, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Acosta, E.P.; Balfour, H.H., Jr. Acyclovir for treatment of postherpetic neuralgia: Efficacy and pharmacokinetics. Antimicrob. Agents Chemother. 2001, 45, 2771–2774. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Watson, C.P. Herpes zoster and postherpetic neuralgia: Past, present and future. Pain Res. Manag. 2009, 14, 275–282. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Semionov, V.; Shvartzman, P. Post herpetic itching—A treatment dilemma. Clin. J. Pain 2008, 24, 366–368. [Google Scholar] [CrossRef]
- Haberthur, K.; Messaoudi, I. Animal models of varicella zoster virus infection. Pathogens 2013, 2, 364–382. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Carpenter, J.E.; Buckingham, E.M.; Jackson, W.; Knudtson, K.; Moffat, J.F.; Kita, H.; Grose, C. Cellular Stress Response to Varicella-Zoster Virus Infection of Human Skin Includes Highly Elevated Interleukin-6 Expression. Open Forum. Infect. Dis. 2018, 5, ofy118. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laval, K.; Enquist, L.W. The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens 2020, 9, 254. https://doi.org/10.3390/pathogens9040254
Laval K, Enquist LW. The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens. 2020; 9(4):254. https://doi.org/10.3390/pathogens9040254
Chicago/Turabian StyleLaval, Kathlyn, and Lynn W. Enquist. 2020. "The Neuropathic Itch Caused by Pseudorabies Virus" Pathogens 9, no. 4: 254. https://doi.org/10.3390/pathogens9040254
APA StyleLaval, K., & Enquist, L. W. (2020). The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens, 9(4), 254. https://doi.org/10.3390/pathogens9040254