Contribution of Epstein–Barr Virus Latent Proteins to the Pathogenesis of Classical Hodgkin Lymphoma


{kind=link}
{kind=link}
Abstract
1. EBV Is a Transforming B Lymphotropic Virus
2. Asymptomatic Infection of B Cells
3. Hodgkin Lymphoma (HL)
3.1. B-Cell Origin of Hodgkin Lymphoma
3.2. Deregulated Cellular Signalling in Classical Hodgkin Lymphoma
3.3. EBV Is Involved in the Pathogenesis of a Subset of Classical Hodgkin Lymphoma
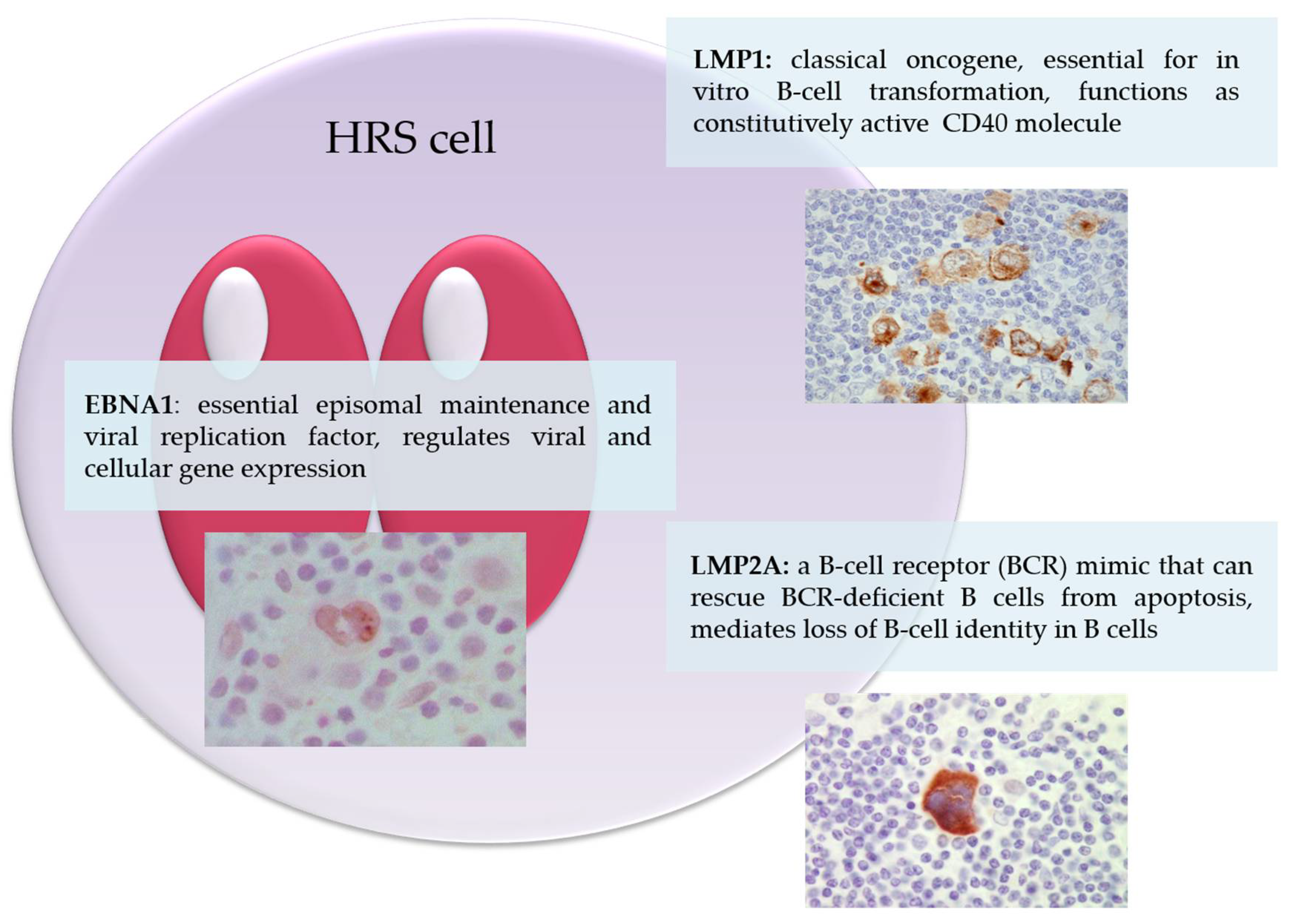
3.4. Contribution of EBV Latent Genes to the Pathogenesis of Classical Hodgkin Lymphoma
3.4.1. Epstein–Barr Virus Nuclear Antigen-1 (EBNA1)
3.4.2. Latent Membrane Protein-1 (LMP1)
3.4.3. Latent Membrane Protein-2 (LMP2)
3.4.4. Potential Interactions between LMP1 and LMP2A in B-Cell Lymphomagenesis
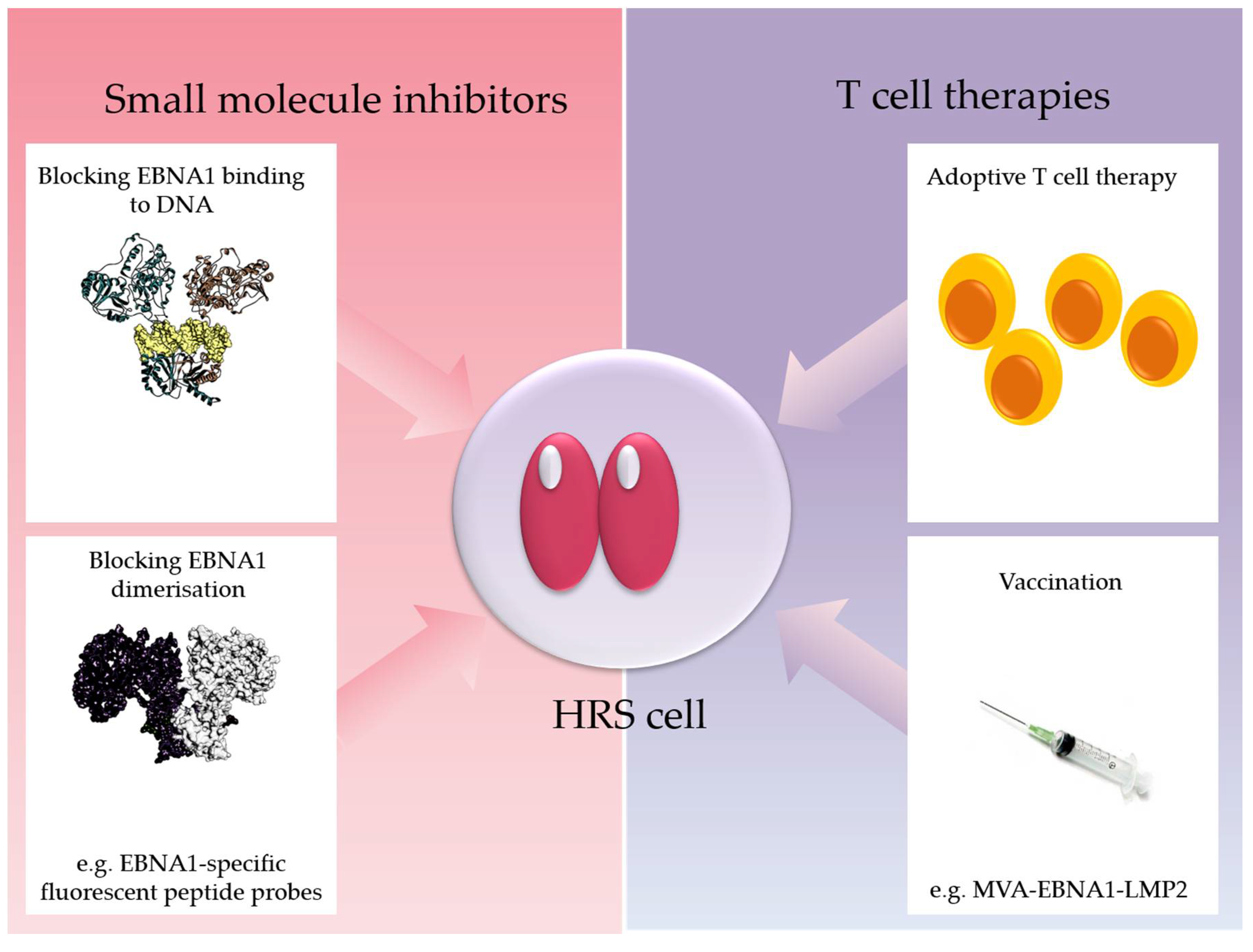
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rickinson, A.B.; Rowe, M.; Hart, I.J.; Yao, Q.Y.; Henderson, L.E.; Rabin, H.; Epstein, M.A. T-cell-mediated regression of “spontaneous” and of epstein-barr virus-induced b-cell transformation in vitro: Studies with cyclosporin a. Cell. Immunol. 1984, 87, 646–658. [Google Scholar] [CrossRef]
- Kerr, B.M.; Lear, A.L.; Rowe, M.; Croom-Carter, D.; Young, L.S.; Rookes, S.M.; Gallimore, P.H.; Rickinson, A.B. Three transcriptionally distinct forms of epstein-barr virus latency in somatic cell hybrids: Cell phenotype dependence of virus promoter usage. Virology 1992, 187, 189–201. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded micrornas. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-barr virus nuclear antigen ebna-lp is essential for transforming naive b cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. Ebv persistence in memory b cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The expression pattern of epstein-barr virus latent genes in vivo is dependent upon the differentiation stage of the infected b cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of epstein-barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-barr virus lmp2a drives b cell development and survival in the absence of normal b cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef]
- Rovedo, M.; Longnecker, R. Epstein-barr virus latent membrane protein 2b (lmp2b) modulates lmp2a activity. J. Virol. 2007, 81, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of epstein-barr virus in vivo. J. Virol. 2004, 79, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sattarzadeh, A.; Diepstra, A.; Visser, L.; van den Berg, A. The microenvironment in classical hodgkin lymphoma: An actively shaped and essential tumor component. Semin. Cancer Biol. 2014, 24, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, I.; Hansmann, M.L.; Franssila, K.; Harris, M.; Harris, N.L.; Jaffe, E.S.; Han, J.; van Krieken, J.M.; Poppema, S.; Marafioti, T.; et al. European task force on lymphoma project on lymphocyte predominance hodgkin disease: Histologic and immunohistologic analysis of submitted cases reveals 2 types of hodgkin disease with a nodular growth pattern and abundant lymphocytes. Blood 2000, 96, 1889–1899. [Google Scholar] [PubMed]
- Kuppers, R.; Rajewsky, K. The origin of hodgkin and reed/sternberg cells in hodgkin’s disease. Annu. Rev. Immunol. 1998, 16, 471–493. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and reed-sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from b cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin’s disease: Clonal ig gene rearrangements in hodgkin and reed-sternberg cells picked from histological sections. Ann. N. Y. Acad. Sci. 1995, 764, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Marafioti, T.; Hummel, M.; Anagnostopoulos, I.; Foss, H.D.; Falini, B.; Delsol, G.; Isaacson, P.G.; Pileri, S.; Stein, H. Origin of nodular lymphocyte-predominant hodgkin’s disease from a clonal expansion of highly mutated germinal-center b cells. N. Engl. J. Med. 1997, 337, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ushmorov, A.; Ritz, O.; Hummel, M.; Leithauser, F.; Moller, P.; Stein, H.; Wirth, T. Epigenetic silencing of the immunoglobulin heavy-chain gene in classical hodgkin lymphoma-derived cell lines contributes to the loss of immunoglobulin expression. Blood 2004, 104, 3326–3334. [Google Scholar] [CrossRef] [PubMed]
- Hertel, C.B.; Zhou, X.G.; Hamilton-Dutoit, S.J.; Junker, S. Loss of b cell identity correlates with loss of b cell-specific transcription factors in hodgkin/reed-sternberg cells of classical hodgkin lymphoma. Oncogene 2002, 21, 4908–4920. [Google Scholar] [CrossRef] [PubMed]
- Schwering, I.; Brauninger, A.; Klein, U.; Jungnickel, B.; Tinguely, M.; Diehl, V.; Hansmann, M.L.; Dalla-Favera, R.; Rajewsky, K.; Kuppers, R. Loss of the b-lineage-specific gene expression program in hodgkin and reed-sternberg cells of hodgkin lymphoma. Blood 2003, 101, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Klein, U.; Schwering, I.; Distler, V.; Brauninger, A.; Cattoretti, G.; Tu, Y.; Stolovitzky, G.A.; Califano, A.; Hansmann, M.L.; et al. Identification of hodgkin and reed-sternberg cell-specific genes by gene expression profiling. J. Clin. Investig. 2003, 111, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Doring, C.; Brune, V.; van Noesel, C.J.; Klapper, W.; Mechtersheimer, G.; Falini, B.; Kuppers, R.; Hansmann, M.L. Analyzing primary hodgkin and reed-sternberg cells to capture the molecular and cellular pathogenesis of classical hodgkin lymphoma. Blood 2012, 120, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Diepstra, A.; Lee, T.; Chan, F.C.; Farinha, P.; Tan, K.; Telenius, A.; Barclay, L.; Shah, S.P.; Connors, J.M.; et al. Gene expression profiling of microdissected hodgkin reed-sternberg cells correlates with treatment outcome in classical hodgkin lymphoma. Blood 2012, 120, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.C.; Leng, C.; Krappmann, D.; Emmerich, F.; Mapara, M.Y.; Bommert, K.; Royer, H.D.; Scheidereit, C.; Dorken, B. High-level nuclear nf-kappa b and oct-2 is a common feature of cultured hodgkin/reed-sternberg cells. Blood 1996, 87, 4340–4347. [Google Scholar] [PubMed]
- Carbone, A.; Gloghini, A.; Gattei, V.; Aldinucci, D.; Degan, M.; De Paoli, P.; Zagonel, V.; Pinto, A. Expression of functional cd40 antigen on reed-sternberg cells and hodgkin’s disease cell lines. Blood 1995, 85, 780–789. [Google Scholar] [PubMed]
- Fiumara, P.; Snell, V.; Li, Y.; Mukhopadhyay, A.; Younes, M.; Gillenwater, A.M.; Cabanillas, F.; Aggarwal, B.B.; Younes, A. Functional expression of receptor activator of nuclear factor kappab in hodgkin disease cell lines. Blood 2001, 98, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Horie, R.; Watanabe, T.; Morishita, Y.; Ito, K.; Ishida, T.; Kanegae, Y.; Saito, I.; Higashihara, M.; Mori, S.; Kadin, M.E.; et al. Ligand-independent signaling by overexpressed cd30 drives nf-kappab activation in hodgkin-reed-sternberg cells. Oncogene 2002, 21, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.; Xu, W.; He, B.; Dillon, S.R.; Gross, J.A.; Sievers, E.; Qiao, X.; Santini, P.; Hyjek, E.; Lee, J.W.; et al. Hodgkin lymphoma cells express taci and bcma receptors and generate survival and proliferation signals in response to baff and april. Blood 2007, 109, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Gruss, H.J.; Pinto, A. Cd40 ligand is constitutively expressed in a subset of t cell lymphomas and on the microenvironmental reactive t cells of follicular lymphomas and hodgkin’s disease. Am. J. Pathol. 1995, 147, 912–922. [Google Scholar] [PubMed]
- Pinto, A.; Aldinucci, D.; Gloghini, A.; Zagonel, V.; Degan, M.; Perin, V.; Todesco, M.; De Iuliis, A.; Improta, S.; Sacco, C.; et al. The role of eosinophils in the pathobiology of hodgkin’s disease. Ann. Oncol. 1997, 8 (Suppl. 2), 89–96. [Google Scholar] [CrossRef] [PubMed]
- Kreher, S.; Bouhlel, M.A.; Cauchy, P.; Lamprecht, B.; Li, S.; Grau, M.; Hummel, F.; Kochert, K.; Anagnostopoulos, I.; Johrens, K.; et al. Mapping of transcription factor motifs in active chromatin identifies irf5 as key regulator in classical hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E4513–E4522. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Dorken, B.; Jundt, F. Notch is an essential upstream regulator of nf-kappab and is relevant for survival of hodgkin and reed-sternberg cells. Leukemia 2012, 26, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.F.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Mooller, P. Gains of 2p involving the rel locus correlate with nuclear c-rel protein accumulation in neoplastic cells of classical hodgkin lymphoma. Blood 2003, 101, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Granzow, M.; Holtgreve-Grez, H.; Siebert, R.; Harder, L.; Martin-Subero, J.I.; Wolf, J.; Adamowicz, M.; Barth, T.F.; Lichter, P.; et al. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including rel and jak2. Int. J. Cancer 2003, 103, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the rel and bcl11a loci in classical hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Telenius, A.; Shah, S.P.; Farinha, P.; Barclay, L.; Boyle, M.; Connors, J.M.; Horsman, D.E.; Gascoyne, R.D. Genome-wide copy number analysis of hodgkin reed-sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 2010, 116, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Cabannes, E.; Khan, G.; Aillet, F.; Jarrett, R.F.; Hay, R.T. Mutations in the ikba gene in hodgkin’s disease suggest a tumour suppressor role for ikappabalpha. Oncogene 1999, 18, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, F.; Meiser, M.; Hummel, M.; Demel, G.; Foss, H.D.; Jundt, F.; Mathas, S.; Krappmann, D.; Scheidereit, C.; Stein, H.; et al. Overexpression of i kappa b alpha without inhibition of nf-kappab activity and mutations in the i kappa b alpha gene in reed-sternberg cells. Blood 1999, 94, 3129–3134. [Google Scholar] [PubMed]
- Jungnickel, B.; Staratschek-Jox, A.; Brauninger, A.; Spieker, T.; Wolf, J.; Diehl, V.; Hansmann, M.L.; Rajewsky, K.; Kuppers, R. Clonal deleterious mutations in the ikappabalpha gene in the malignant cells in hodgkin’s lymphoma. J. Exp. Med. 2000, 191, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, F.; Theurich, S.; Hummel, M.; Haeffker, A.; Vry, M.S.; Dohner, K.; Bommert, K.; Stein, H.; Dorken, B. Inactivating i kappa b epsilon mutations in hodgkin/reed-sternberg cells. J. Pathol. 2003, 201, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.; Shield, L.A.; Cordano, P.; Chui, D.T.; Osborne, J.; Crae, S.; Wilson, K.S.; Tosi, S.; Knight, S.J.; Gesk, S.; et al. Mutations of nfkbia, encoding ikappab alpha, are a recurrent finding in classical hodgkin lymphoma but are not a unifying feature of non-ebv-associated cases. Int. J. Cancer 2009, 125, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Wlodarska, I.; Bastard, C.; Picquenot, J.M.; Hoppner, J.; Giefing, M.; Klapper, W.; Siebert, R. Chromosomal rearrangements involving the bcl3 locus are recurrent in classical hodgkin and peripheral t-cell lymphoma. Blood 2006, 108, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Johrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P.; et al. Elevated nf-kappab p50 complex formation and bcl-3 expression in classical hodgkin, anaplastic large-cell, and other peripheral t-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. Tnfaip3 (a20) is a tumor suppressor gene in hodgkin lymphoma and primary mediastinal b cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary hodgkin and reed-sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Pittaluga, S.; Evbuomwan, M.O.; Jaffe, E.S.; Lewis, B.A. Hodgkin lymphoma requires stabilized nik and constitutive relb expression for survival. Blood 2012, 120, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Giefing, M.; Massow, A.; Vater, I.; Gesk, S.; Schlesner, M.; Richter, J.; Klapper, W.; Hansmann, M.L.; Siebert, R.; et al. Genetic lesions of the traf3 and map3k14 genes in classical hodgkin lymphoma. Br. J. Haematol. 2012, 157, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, L.; Gloghini, A.; Olivo, K.; Di Francia, R.; Lorenzon, D.; De Filippi, R.; Carbone, A.; Colombatti, A.; Pinto, A.; Aldinucci, D. Functional coexpression of interleukin (il)-7 and its receptor (il-7r) on hodgkin and reed-sternberg cells: Involvement of il-7 in tumor cell growth and microenvironmental interactions of hodgkin’s lymphoma. Int. J. Cancer 2009, 125, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, B.F.; Elia, A.J.; Gascoyne, R.D.; Trumper, L.H.; von Bonin, F.; Kapp, U.; Patterson, B.; Snow, B.E.; Mak, T.W. Interleukin 13 and interleukin 13 receptor are frequently expressed by hodgkin and reed-sternberg cells of hodgkin lymphoma. Blood 2001, 97, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Kapp, U.; Yeh, W.-C.; Patterson, B.; Elia, A.J.; Kägi, D.; Ho, A.; Hessel, A.; Tipsword, M.; Williams, A.; Mirtsos, C.; et al. Interleukin 13 is secreted by and stimulates the growth of hodgkin and reed-sternberg cells. J. Exp. Med. 1999, 189, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Gruss, H.J.; Brach, M.A.; Drexler, H.G.; Bross, K.J.; Herrmann, F. Interleukin-9 is expressed by primary and cultured hodgkin and reed-sternberg cells. Cancer Res. 1992, 52, 1026–1031. [Google Scholar] [PubMed]
- Aldinucci, D.; Poletto, D.; Gloghini, A.; Nanni, P.; Degan, M.; Perin, T.; Ceolin, P.; Rossi, F.M.; Gattei, V.; Carbone, A.; et al. Expression of functional interleukin-3 receptors on hodgkin and reed-sternberg cells. Am. J. Pathol. 2002, 160, 585–596. [Google Scholar] [CrossRef]
- Lamprecht, B.; Kreher, S.; Anagnostopoulos, I.; Johrens, K.; Monteleone, G.; Jundt, F.; Stein, H.; Janz, M.; Dorken, B.; Mathas, S. Aberrant expression of the th2 cytokine il-21 in hodgkin lymphoma cells regulates stat3 signaling and attracts treg cells via regulation of mip-3alpha. Blood 2008, 112, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.; et al. Il-21 is expressed in hodgkin lymphoma and activates stat5: Evidence that activated stat5 is required for hodgkin lymphomagenesis. Blood 2008, 111, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Lemke, P.; Anagnostopoulos, I.; Hacker, C.; Krappmann, D.; Mathas, S.; Dörken, B.; Zenke, M.; Stein, H.; Scheidereit, C. Nuclear factor κb–dependent gene expression profiling of hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J. Exp. Med. 2002, 196, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, B.F.; Elia, A.J.; Gascoyne, R.D.; Patterson, B.; Trumper, L.; Kapp, U.; Mak, T.W. Signal transducer and activator of transcription 6 is frequently activated in hodgkin and reed-sternberg cells of hodgkin lymphoma. Blood 2002, 99, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Holtick, U.; Vockerodt, M.; Ahmadi, T.; Haier, B.; Behrmann, I.; Heinrich, P.C.; Diehl, V.; Tesch, H. Stat3 is constitutively activated in hodgkin cell lines. Blood 2001, 98, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Kupper, M.; Ohl, S.; von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Moller, P.; Pfreundschuh, M.; Trumper, L.; et al. Genomic imbalances including amplification of the tyrosine kinase gene jak2 in cd30+ hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Weniger, M.A.; Melzner, I.; Menz, C.K.; Wegener, S.; Bucur, A.J.; Dorsch, K.; Mattfeldt, T.; Barth, T.F.; Moller, P. Mutations of the tumor suppressor gene socs-1 in classical hodgkin lymphoma are frequent and associated with nuclear phospho-stat5 accumulation. Oncogene 2006, 25, 2679–2684. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, J.; Chan, F.C.; Telenius, A.; Woolcock, B.; Kridel, R.; Tan, K.L.; Ben-Neriah, S.; Mottok, A.; Lim, R.S.; Boyle, M.; et al. Recurrent somatic mutations of ptpn1 in primary mediastinal b cell lymphoma and hodgkin lymphoma. Nat. Genet. 2014, 46, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Penson, A.; Schiavoni, G.; Ladewig, E.; Fortini, E.; Wang, Y.C.; Spanhol-Rosseto, A.; Venanzi, A.; Gianni, A.M.; Viviani, S.; et al. New recurrently mutated genes in classical hodgkin lymphoma revealed by whole-exome sequencing of microdissected tumor cells. Blood 2016, 128, 1088. [Google Scholar]
- Zahn, M.; Marienfeld, R.; Melzner, I.; Heinrich, J.; Renner, B.; Wegener, S.; Miessner, A.; Barth, T.F.; Dorsch, K.; Bruderlein, S.; et al. A novel ptpn1 splice variant upregulates jak/stat activity in classical hodgkin lymphoma cells. Blood 2017, 129, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiss, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.L.; Kuppers, R.; et al. An oncogenic axis of stat-mediated batf3 upregulation causing myc activity in classical hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dorken, B.; et al. Aberrantly expressed c-jun and junb are a hallmark of hodgkin lymphoma cells, stimulate proliferation and synergize with nf-kappa b. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.; Reynolds, G.M.; Dawson, C.W.; Young, L.S.; Murray, P.G. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of hodgkin’s lymphoma cells through a mechanism involving akt kinase and mtor. J. Pathol. 2005, 205, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, G.V.; Li, Y.; Rassidakis, G.Z.; Medeiros, L.J.; Mills, G.B.; Younes, A. Inhibition of the phosphatidylinositol-3 kinase/akt promotes g1 cell cycle arrest and apoptosis in hodgkin lymphoma. Br. J. Haematol. 2006, 132, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Vrzalikova, K.; Ibrahim, M.; Vockerodt, M.; Perry, T.; Margielewska, S.; Lupino, L.; Nagy, E.; Soilleux, E.; Liebelt, D.; Hollows, R.; et al. S1pr1 drives a feed forward signalling loop to regulate batf3 and the transcriptional programme of hodgkin lymphoma cells. Leukemia 2017, 32, 214. [Google Scholar] [CrossRef] [PubMed]
- Willenbrock, K.; Kuppers, R.; Renne, C.; Brune, V.; Eckerle, S.; Weidmann, E.; Brauninger, A.; Hansmann, M.L. Common features and differences in the transcriptome of large cell anaplastic lymphoma and classical hodgkin’s lymphoma. Haematologica 2006, 91, 596–604. [Google Scholar] [PubMed]
- Renne, C.; Minner, S.; Kuppers, R.; Hansmann, M.L.; Brauninger, A. Autocrine ngfbeta/trka signalling is an important survival factor for hodgkin lymphoma derived cell lines. Leuk. Res. 2008, 32, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Renne, C.; Willenbrock, K.; Kuppers, R.; Hansmann, M.L.; Brauninger, A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic hodgkin lymphoma. Blood 2005, 105, 4051–4059. [Google Scholar] [CrossRef] [PubMed]
- Cader, F.Z.; Vockerodt, M.; Bose, S.; Nagy, E.; Brundler, M.A.; Kearns, P.; Murray, P.G. The ebv oncogene lmp1 protects lymphoma cells from cell death through the collagen-mediated activation of ddr1. Blood 2013, 122, 4237–4245. [Google Scholar] [CrossRef] [PubMed]
- Levine, P.H.; Ablashi, D.V.; Berard, C.W.; Carbone, P.P.; Waggoner, D.E.; Malan, L. Elevated antibody titers to epstein-barr virus in hodgkin’s disease. Cancer 1971, 27, 416–421. [Google Scholar] [CrossRef]
- Mueller, N.; Evans, A.; Harris, N.L.; Comstock, G.W.; Jellum, E.; Magnus, K.; Orentreich, N.; Polk, B.F.; Vogelman, J. Hodgkin’s disease and epstein-barr virus. Altered antibody pattern before diagnosis. N. Engl. J. Med. 1989, 320, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Connelly, R.R.; Christine, B.W. A cohort study of cancer following infectious mononucleosis. Cancer Res. 1974, 34, 1172–1178. [Google Scholar] [PubMed]
- Rosdahl, N.; Larsen, S.O.; Clemmesen, J. Hodgkin’s disease in patients with previous infectious mononucleosis: 30 years’ experience. BMJ 1974, 2, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Hjalgrim, H.; Smedby, K.E.; Rostgaard, K.; Molin, D.; Hamilton-Dutoit, S.; Chang, E.T.; Ralfkiaer, E.; Sundstrom, C.; Adami, H.O.; Glimelius, B.; et al. Infectious mononucleosis, childhood social environment, and risk of hodgkin lymphoma. Cancer Res. 2007, 67, 2382–2388. [Google Scholar] [CrossRef] [PubMed]
- Hjalgrim, H.; Munksgaard, L.; Melbye, M. Epstein-barr virus and hodgkin’s lymphoma. Ugeskr Laeger 2002, 164, 5924–5927. [Google Scholar] [PubMed]
- Poppema, S.; van Imhoff, G.; Torensma, R.; Smit, J. Lymphadenopathy morphologically consistent with hodgkin’s disease associated with epstein-barr virus infection. Am. J. Clin. Pathol. 1985, 84, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.M.; Strickler, J.G.; Warnke, R.A.; Purtilo, D.T.; Sklar, J. Epstein-barr viral DNA in tissues of hodgkin’s disease. Am. J. Pathol. 1987, 129, 86–91. [Google Scholar] [PubMed]
- Anagnostopoulos, I.; Herbst, H.; Niedobitek, G.; Stein, H. Demonstration of monoclonal ebv genomes in hodgkin’s disease and ki-1-positive anaplastic large cell lymphoma by combined southern blot and in situ hybridization. Blood 1989, 74, 810–816. [Google Scholar] [PubMed]
- Weiss, L.M.; Movahed, L.A.; Warnke, R.A.; Sklar, J. Detection of epstein-barr viral genomes in reed-sternberg cells of hodgkin’s disease. N. Engl. J. Med. 1989, 320, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-C.; Mann, R.B.; Charache, P.; Hayward, S.D.; Staal, S.; Lambe, B.C.; Ambinder, R.F. Detection of ebv gene expression in reed-sternberg cells of hodgkin’s disease. Int. J. Cancer 1990, 46, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Coates, P.J.; Slavin, G.; D’Ardenne, A.J. Persistence of epstein-barr virus in reed-sternberg cells throughout the course of hodgkin’s disease. J. Pathol. 1991, 164, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.L.; Lin, R.J.; Stewart, S.L.; Ambinder, R.F.; Jarrett, R.F.; Brousset, P.; Pallesen, G.; Gulley, M.L.; Khan, G.; O’Grady, J.; et al. Epstein-barr virus-associated hodgkin’s disease: Epidemiologic characteristics in international data. Int. J. Cancer 1997, 70, 375–382. [Google Scholar] [CrossRef]
- Glaser, S.L.; Jarrett, R.F. The epidemiology of Hodgkin’s disease. Baillieres Clin. Haematol. 1996, 9, 401–416. [Google Scholar] [CrossRef]
- Chang, K.L.; Albujar, P.F.; Chen, Y.Y.; Johnson, R.M.; Weiss, L.M. High prevalence of epstein-barr virus in the reed-sternberg cells of hodgkin’s disease occurring in peru. Blood 1993, 81, 496–501. [Google Scholar] [PubMed]
- Weinreb, M.; Day, P.J.; Niggli, F.; Green, E.K.; Nyong’o, A.O.; Othieno-Abinya, N.A.; Riyat, M.S.; Raafat, F.; Mann, J.R. The consistent association between epstein-barr virus and hodgkin’s disease in children in kenya. Blood 1996, 87, 3828–3836. [Google Scholar] [PubMed]
- Armstrong, A.A.; Alexander, F.E.; Cartwright, R.; Angus, B.; Krajewski, A.S.; Wright, D.H.; Brown, I.; Lee, F.; Kane, E.; Jarrett, R.F. Epstein-barr virus and hodgkin’s disease: Further evidence for the three disease hypothesis. Leukemia 1998, 12, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, R.F.; Gallagher, A.; Jones, D.B.; Alexander, F.E.; Krajewski, A.S.; Kelsey, A.; Adams, J.; Angus, B.; Gledhill, S.; Wright, D.H.; et al. Detection of epstein-barr virus genomes in hodgkin’s disease: Relation to age. J. Clin. Pathol. 1991, 44, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Flavell, K.J.; Biddulph, J.P.; Powell, J.E.; Parkes, S.E.; Redfern, D.; Weinreb, M.; Nelson, P.; Mann, J.R.; Young, L.S.; Murray, P.G. South asian ethnicity and material deprivation increase the risk of epstein-barr virus infection in childhood hodgkin’s disease. Br. J. Cancer 2001, 85, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Westhoff Smith, D.; Sugden, B. Potential cellular functions of epstein-barr nuclear antigen 1 (ebna1) of epstein-barr virus. Viruses 2013, 5, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. Contributions of epstein-barr nuclear antigen 1 (ebna1) to cell immortalization and survival. Viruses 2012, 4, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. Ebna1 and host factors in epstein-barr virus latent DNA replication. Curr. Opin. Virol. 2012, 2, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. The epstein-barr virus ebna1 protein. Scientifica (Cairo) 2012, 2012, 438204. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-barr virus provides a survival factor to burkitt’s lymphomas. Proc. Natl. Acad. Sci. USA 2003, 100, 14269–14274. [Google Scholar] [CrossRef] [PubMed]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of hausp/usp7 bound to epstein-barr nuclear antigen 1 implications for ebv-mediated immortalization. Mol. Cell. 2005, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Vockerodt, M.; Weber, O.; Hell, K.; Wolf, J.; Haier, B.; Grasser, F.A.; Muller-Lantzsch, N.; Kieff, E.; Diehl, V.; et al. Expression of epstein-barr virus nuclear antigen 1 is associated with enhanced expression of cd25 in the hodgkin cell line l428. J. Virol. 1999, 73, 1630–1636. [Google Scholar] [PubMed]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the tgf-beta target gene, ptprk, by the epstein-barr virus encoded ebna1 contributes to the growth and survival of hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.H.; O’Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-barr virus-encoded ebna1 regulates cellular gene transcription and modulates the stat1 and tgfbeta signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Baumforth, K.R.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the epstein-barr virus-encoded epstein-barr virus nuclear antigen 1 in hodgkin’s lymphoma cells mediates up-regulation of ccl20 and the migration of regulatory t cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of epstein-barr virus nuclear antigen-1 induces b cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar] [PubMed]
- Tsimbouri, P.; Drotar, M.E.; Coy, J.L.; Wilson, J.B. Bcl-xl and rag genes are induced and the response to il-2 enhanced in emuebna-1 transgenic mouse lymphocytes. Oncogene 2002, 21, 5182–5187. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Lu, H.; Yasui, T.; Sharpe, A.; Warren, H.; Cahir-McFarland, E.; Bronson, R.; Hung, S.C.; Kieff, E. Epstein-barr virus nuclear antigen 1 does not induce lymphoma in transgenic fvb mice. Proc. Natl. Acad. Sci. USA 2005, 102, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Coppotelli, G.; Mughal, N.; Callegari, S.; Sompallae, R.; Caja, L.; Luijsterburg, M.S.; Dantuma, N.P.; Moustakas, A.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group a proteins. Nucleic Acids Res. 2013, 41, 2950–2962. [Google Scholar] [CrossRef] [PubMed]
- Dresang, L.R.; Vereide, D.T.; Sugden, B. Identifying sites bound by epstein-barr virus nuclear antigen 1 (ebna1) in the human genome: Defining a position-weighted matrix to predict sites bound by ebna1 in viral genomes. J. Virol. 2009, 83, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wikramasinghe, P.; Norseen, J.; Tsai, K.; Wang, P.; Showe, L.; Davuluri, R.V.; Lieberman, P.M. Genome-wide analysis of host-chromosome binding sites for epstein-barr virus nuclear antigen 1 (ebna1). Virol. J. 2010, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Canaan, A.; Haviv, I.; Urban, A.E.; Schulz, V.P.; Hartman, S.; Zhang, Z.; Palejev, D.; Deisseroth, A.B.; Lacy, J.; Snyder, M.; et al. Ebna1 regulates cellular gene expression by binding cellular promoters. Proc. Natl. Acad. Sci. USA 2009, 106, 22421–22426. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; De Leo, A.; Kossenkov, A.V.; Cesaroni, M.; Song, H.; Dawany, N.; Showe, L.; Lu, F.; Wikramasinghe, P.; Lieberman, P.M. Identification of mef2b, ebf1, and il6r as direct gene targets of epstein-barr virus (ebv) nuclear antigen 1 critical for ebv-infected b-lymphocyte survival. J. Virol. 2016, 90, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Thompson, S.; Schultz, D.C.; Zhu, W.; Jiang, H.; Luo, C.; Lieberman, P.M. Discovery of selective inhibitors against ebna1 via high throughput in silico virtual screening. PLoS ONE 2010, 5, e10126. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Messick, T.; Schultz, D.C.; Reichman, M.; Lieberman, P.M. Development of a high-throughput screen for inhibitors of epstein-barr virus ebna1. J. Biomol. Screen 2010, 15, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lui, Y.L.; Li, H.; Chan, C.F.; Lan, R.; Chan, W.L.; Lau, T.C.; Tsao, G.S.; Mak, N.K.; Wong, K.L. Ebna1-specific luminescent small molecules for the imaging and inhibition of latent ebv-infected tumor cells. Chem. Commun. 2014, 50, 6517–6519. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding epstein-barr virus (ebv) target antigens: A phase i trial in uk patients with ebv-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Nourse, J.P.; Morrison, L.; Nguyen-Van, D.; Moss, D.J.; Burrows, S.R.; Gandhi, M.K. Expansion of ebna1-specific effector t cells in posttransplantation lymphoproliferative disorders. Blood 2010, 116, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of epstein-barr virus (ebv) nuclear antigen 1-specific t cells as treatment for ebv reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013, 31, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Lista, M.J.; Martins, R.P.; Billant, O.; Contesse, M.A.; Findakly, S.; Pochard, P.; Daskalogianni, C.; Beauvineau, C.; Guetta, C.; Jamin, C.; et al. Nucleolin directly mediates epstein-barr virus immune evasion through binding to g-quadruplexes of ebna1 mrna. Nat. Commun. 2017, 8, 16043. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.; Sugden, B. Cd40 and its viral mimic, lmp1: Similar means to different ends. Cell. Signal. 2003, 15, 9–16. [Google Scholar] [CrossRef]
- Kaykas, A.; Worringer, K.; Sugden, B. Cd40 and lmp-1 both signal from lipid rafts but lmp-1 assembles a distinct, more efficient signaling complex. Embo J. 2001, 20, 2641–2654. [Google Scholar] [CrossRef] [PubMed]
- Bishop, G.A.; Hostager, B.S. Signaling by cd40 and its mimics in b cell activation. Immunol. Res. 2001, 24, 97–110. [Google Scholar] [CrossRef]
- Panagopoulos, D.; Victoratos, P.; Alexiou, M.; Kollias, G.; Mosialos, G. Comparative analysis of signal transduction by cd40 and the epstein-barr virus oncoprotein lmp1 in vivo. J. Virol. 2004, 78, 13253–13261. [Google Scholar] [CrossRef] [PubMed]
- Kilger, E.; Kieser, A.; Baumann, M.; Hammerschmidt, W. Epstein-barr virus-mediated b-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated cd40 receptor. EMBO J. 1998, 17, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Kohlhuber, F.; Kilger, E.; Baumann, M.; Kieser, A.; Kaiser, C.; Zeidler, R.; Scheffer, B.; Ueffing, M.; Hammerschmidt, W. Latent membrane protein 1 of epstein-barr virus interacts with jak3 and activates stat proteins. EMBO J. 1999, 18, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Young, L.S. Activation of the cjun n-terminal kinase (jnk) pathway by the epstein-barr virus-encoded latent membrane protein 1 (lmp1). Oncogene 1998, 16, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Laherty, C.D.; Hu, H.M.; Opipari, A.W.; Wang, F.; Dixit, V.M. The epstein-barr virus lmp1 gene product induces a20 zinc finger protein expression by activating nuclear factor kappa b. J. Biol. Chem. 1992, 267, 24157–24160. [Google Scholar] [PubMed]
- Huen, D.S.; Henderson, S.A.; Croom-Carter, D.; Rowe, M. The epstein-barr virus latent membrane protein-1 (lmp1) mediates activation of nf-kappa b and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995, 10, 549–560. [Google Scholar] [PubMed]
- Schumacher, M.A.; Schmitz, R.; Brune, V.; Tiacci, E.; Doring, C.; Hansmann, M.L.; Siebert, R.; Kuppers, R. Mutations in the genes coding for the nf-kappab regulating factors ikappabalpha and a20 are uncommon in nodular lymphocyte-predominant hodgkin’s lymphoma. Haematologica 2010, 95, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Etzel, B.M.; Gerth, M.; Chen, Y.; Wunsche, E.; Facklam, T.; Beck, J.F.; Guntinas-Lichius, O.; Petersen, I. Mutation analysis of tumor necrosis factor alpha-induced protein 3 gene in hodgkin lymphoma. Pathol. Res. Pract. 2017, 213, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Morgan, S.L.; Kuo, M.; Wei, W.; Chukwuma, M.B.; Arrand, J.R.; Kube, D.; Gordon, J.; Young, L.S.; Woodman, C.B.; et al. The epstein-barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre b cells towards a hodgkin’s reed-sternberg-like phenotype. J. Pathol. 2008, 216, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.; Rowe, M.; Murray, P.G.; et al. The h3k27me3 demethylase, kdm6b, is induced by epstein-barr virus and over-expressed in hodgkin’s lymphoma. Oncogene 2011, 30, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Gordon, N.; Smith, N.; Rowe, M.; Murray, P.G.; Woodman, C.B. Arginine methyltransferases are regulated by epstein-barr virus in b cells and are differentially expressed in hodgkin’s lymphoma. Pathogens 2012, 1, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Wei, W.; Anderton, J.; Vockerodt, M.; Rowe, M.; Murray, P.G.; Woodman, C.B. Epigenetic and transcriptional changes which follow epstein-barr virus infection of germinal center b cells and their relevance to the pathogenesis of hodgkin’s lymphoma. J. Virol. 2011, 85, 9568–9577. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.A.; Lupey, L.N.; Tempera, I. Epstein-barr virus oncoprotein lmp1 mediates epigenetic changes in host gene expression through parp1. J. Virol. 2016, 90, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Motsch, N.; Pfuhl, T.; Mrazek, J.; Barth, S.; Grasser, F.A. Epstein-barr virus-encoded latent membrane protein 1 (lmp1) induces the expression of the cellular microrna mir-146a. RNA Biol. 2007, 4, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.E.; Yin, Q.; Fewell, C.; Lacey, M.; McBride, J.; Wang, X.; Lin, Z.; Schaefer, B.C.; Flemington, E.K. Epstein-barr virus latent membrane protein 1 induces cellular microrna mir-146a, a modulator of lymphocyte signaling pathways. J. Virol. 2008, 82, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Weidmer, A.; Liu, C.G.; Volinia, S.; Croce, C.M.; Lieberman, P.M. Epstein-barr virus-induced mir-155 attenuates nf-kappab signaling and stabilizes latent virus persistence. J. Virol. 2008, 82, 10436–10443. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Boccellato, F.; Vincenti, S.; Rosato, P.; Bozzoni, I.; Frati, L.; Faggioni, A.; Presutti, C.; Trivedi, P. Epstein-barr virus encoded lmp1 downregulates tcl1 oncogene through mir-29b. Oncogene 2010, 29, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, Z.; Peng, Y.; Liu, X.; Lu, J.; Wang, L.; Pan, Q.; He, M.L.; Li, X.P. Microrna-10b induced by epstein-barr virus-encoded latent membrane protein-1 promotes the metastasis of human nasopharyngeal carcinoma cells. Cancer Lett. 2010, 299, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cahir-McFarland, E.D.; Carter, K.; Rosenwald, A.; Giltnane, J.M.; Henrickson, S.E.; Staudt, L.M.; Kieff, E. Role of nf-kappa b in cell survival and transcription of latent membrane protein 1 - expressing or epstein-barr virus latency iii-infected cells. J. Virol. 2004, 78, 4108–4119. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.; O’Neil, J.D.; Milner, A.E.; Reynolds, G.M.; Starczynski, J.; Crocker, J.; Young, L.S.; Murray, P.G. Expression of the cellular flice-inhibitory protein (c-flip) protects hodgkin’s lymphoma cells from autonomous fas-mediated death. Proc. Natl. Acad. Sci. USA 2004, 101, 6611–6616. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, V.; Lemieux, B.; Sawan, B.; Lichtensztejn, D.; Lichtensztejn, Z.; Wellinger, R.; Mai, S.; Knecht, H. Lmp1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 2015, 125, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Mai, S. Lmp1 and dynamic progressive telomere dysfunction: A major culprit in ebv-associated hodgkin’s lymphoma. Viruses 2017, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of blimp1α by the ebv oncogene, lmp-1, disrupts the plasma cell differentiation program and prevents viral replication in b cells: Implications for the pathogenesis of ebv-associated b-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Sueur, C.; Lupo, J.; Mas, P.; Morand, P.; Boyer, V. Difference in cytokine production and cell cycle progression induced by epstein-barr virus lmp1 deletion variants in kmh2, a hodgkin lymphoma cell line. Virol. J. 2014, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Takahara, M.; Nagy, N.; Klein, G.; Klein, E. Cytokine mediated induction of the major epstein-barr virus (ebv)-encoded transforming protein, lmp-1. Immunol. Lett. 2006, 104, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Dukers, D.F.; Jaspars, L.H.; Vos, W.; Oudejans, J.J.; Hayes, D.; Cillessen, S.; Middeldorp, J.M.; Meijer, C.J. Quantitative immunohistochemical analysis of cytokine profiles in epstein-barr virus-positive and -negative cases of hodgkin’s disease. J. Pathol. 2000, 190, 143–149. [Google Scholar] [CrossRef]
- Rickinson, A.B.; Moss, D.J. Human cytotoxic t lymphocyte responses to epstein-barr virus infection. Annu. Rev. Immunol. 1997, 15, 405–431. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.L.; Munz, C. Cytolytic t lymphocytes from hla-b8+ donors frequently recognize the hodgkin’s lymphoma associated latent membrane protein 2 of epstein barr virus. Herpesviridae 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Gottschalk, S.; Huls, M.H.; Molldrem, J.; Przepiorka, D.; Rooney, C.M.; Heslop, H.E. In vivo expansion of lmp 1- and 2-specific t-cells in a patient who received donor-derived ebv-specific t-cells after allogeneic stem cell transplantation. Leuk. Lymphoma 2006, 47, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Sherritt, M.; Thomson, S.; Tellam, J.; Cooper, L.; Connolly, G.; Bharadwaj, M.; Khanna, R. Therapeutic lmp1 polyepitope vaccine for ebv-associated hodgkin disease and nasopharyngeal carcinoma. Blood 2003, 101, 3150–3156. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Edwards, O.L.; Sili, U.; Huls, M.H.; Goltsova, T.; Davis, A.R.; Heslop, H.E.; Rooney, C.M. Generating ctls against the subdominant epstein-barr virus lmp1 antigen for the adoptive immunotherapy of ebv-associated malignancies. Blood 2003, 101, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.L.; Rickinson, A.B.; Thomas, W.A.; Jarrett, R.F.; Crocker, J.; Lee, S.P. Epstein-barr virus-specific cytotoxic t lymphocyte responses in the blood and tumor site of hodgkin’s disease patients: Implications for a t-cell-based therapy. Cancer Res. 2001, 61, 6219–6226. [Google Scholar] [PubMed]
- Khanna, R.; Burrows, S.R.; Nicholls, J.; Poulsen, L.M. Identification of cytotoxic t cell epitopes within epstein-barr virus (ebv) oncogene latent membrane protein 1 (lmp1): Evidence for hla a2 supertype-restricted immune recognition of ebv-infected cells by lmp1-specific cytotoxic t lymphocytes. Eur. J. Immunol. 1998, 28, 451–458. [Google Scholar] [CrossRef]
- Murray, P.G.; Constandinou, C.M.; Crocker, J.; Young, L.S.; Ambinder, R.F. Analysis of major histocompatibility complex class i, tap expression, and lmp2 epitope sequence in epstein-barr virus-positive hodgkin’s disease. Blood 1998, 92, 2477–2483. [Google Scholar] [PubMed]
- Lee, S.P.; Constandinou, C.M.; Thomas, W.A.; Croom-Carter, D.; Blake, N.W.; Murray, P.G.; Crocker, J.; Rickinson, A.B. Antigen presenting phenotype of hodgkin reed-sternberg cells: Analysis of the hla class i processing pathway and the effects of interleukin-10 on epstein-barr virus-specific cytotoxic t-cell recognition. Blood 1998, 92, 1020–1030. [Google Scholar] [PubMed]
- Tsang, C.W.; Lin, X.; Gudgeon, N.H.; Taylor, G.S.; Jia, H.; Hui, E.P.; Chan, A.T.; Lin, C.K.; Rickinson, A.B. Cd4+ t-cell responses to epstein-barr virus nuclear antigen ebna1 in chinese populations are highly focused on novel c-terminal domain-derived epitopes. J. Virol. 2006, 80, 8263–8266. [Google Scholar] [CrossRef] [PubMed]
- Gurer, C.; Strowig, T.; Brilot, F.; Pack, M.; Trumpfheller, C.; Arrey, F.; Park, C.G.; Steinman, R.M.; Munz, C. Targeting the nuclear antigen 1 of epstein-barr virus to the human endocytic receptor dec-205 stimulates protective t-cell responses. Blood 2008, 112, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. Pd-l1 expression is characteristic of a subset of aggressive b-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive ap-1 activity and ebv infection induce pd-l1 in hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased pd-1 ligand expression, and further induction via jak2 in nodular sclerosing hodgkin lymphoma and primary mediastinal large b-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Paydas, S.; Bagir, E.; Seydaoglu, G.; Ercolak, V.; Ergin, M. Programmed death-1 (pd-1), programmed death-ligand 1 (pd-l1), and ebv-encoded rna (eber) expression in hodgkin lymphoma. Ann. Hematol. 2015, 94, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Salamon, D.; Persson, E.K.; Nagy, N.; Scheeren, F.A.; Spits, H.; Klein, G.; Klein, E. Il-21 imposes a type ii ebv gene expression on type iii and type i b cells by the repression of c- and activation of lmp-1-promoter. Proc. Natl. Acad. Sci. USA 2010, 107, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Gerasimcik, N.; Salamon, D.; Persson, E.K.; Nagy, N.; Klein, G.; Severinson, E.; Klein, E. Stat6 signaling pathway activated by the cytokines il-4 and il-13 induces expression of the epstein-barr virus-encoded protein lmp-1 in absence of ebna-2: Implications for the type ii ebv latent gene expression in hodgkin lymphoma. Blood 2011, 117, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Swart, R.; Katzman, R.B.; Ikeda, M.; Ikeda, A.; Longnecker, R.; Dykstra, M.L.; Pierce, S.K. The effects of the epstein-barr virus latent membrane protein 2a on b cell function. Int. Rev. Immunol. 2001, 20, 805–835. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Epstein-barr virus latent membrane protein 2a mediates transformation through constitutive activation of the ras/pi3-k/akt pathway. J. Virol. 2007, 81, 9299–9306. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center b cells from apoptosis by epstein-barr virus. Blood 2005, 106, 4339–4344. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, S.; Bell Ai, A.I.; Pastor, N.B.; Milner, A.E.; Drayson, M.; Gordon, J.; Rickinson, A.B. Epstein-barr virus infection in vitro can rescue germinal center b cells with inactivated immunoglobulin genes. Blood 2005, 106, 4249–4252. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, D.; Kurth, J.; Unkel, C.; Küppers, R. Transformation of bcr-deficient germinal-center b cells bybv supports a major role of the virus in the pathogenesis of hodgkin and posttransplantation lymphomas. Blood 2005, 106, 4345–4350. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-barr virus latent membrane protein 2a is a b-cell receptor mimic and essential for b-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Wei, W.; Nagy, E.; Prouzova, Z.; Schrader, A.; Kube, D.; Rowe, M.; Woodman, C.B.; Murray, P.G. Suppression of the lmp2a target gene, egr-1, protects hodgkin’s lymphoma cells from entry to the ebv lytic cycle. J. Pathol. 2013, 230, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Portis, T.; Longnecker, R. Epstein-barr virus (ebv) lmp2a alters normal transcriptional regulation following b-cell receptor activation. Virology 2004, 318, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Portis, T.; Dyck, P.; Longnecker, R. Epstein-barr virus (ebv) lmp2a induces alterations in gene transcription similar to those observed in reed-sternberg cells of hodgkin lymphoma. Blood 2003, 102, 4166–4178. [Google Scholar] [CrossRef] [PubMed]
- Portis, T.; Longnecker, R. Epstein-barr virus lmp2a interferes with global transcription factor regulation when expressed during b-lymphocyte development. J. Virol. 2003, 77, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Longnecker, R. Epstein-barr virus latent membrane protein 2a exploits notch1 to alter b-cell identity in vivo. Blood 2009, 113, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.A.; Miller, S.D.; Longnecker, R. Epstein-barr virus latent membrane protein 2a exacerbates experimental autoimmune encephalomyelitis and enhances antigen presentation function. Sci. Rep. 2012, 2, 353. [Google Scholar] [CrossRef] [PubMed]
- Kulwichit, W.; Edwards, R.H.; Davenport, E.M.; Baskar, J.F.; Godfrey, V.; Raab-Traub, N. Expression of the epstein-barr virus latent membrane protein 1 induces b cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 11963–11968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kracker, S.; Yasuda, T.; Casola, S.; Vanneman, M.; Homig-Holzel, C.; Wang, Z.; Derudder, E.; Li, S.; Chakraborty, T.; et al. Immune surveillance and therapy of lymphomas driven by epstein-barr virus protein lmp1 in a mouse model. Cell 2012, 148, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Vrazo, A.C.; Chauchard, M.; Raab-Traub, N.; Longnecker, R. Epstein-barr virus lmp2a reduces hyperactivation induced by lmp1 to restore normal b cell phenotype in transgenic mice. PLoS Pathog. 2012, 8, e1002662. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Tsai, M.H.; Romero-Masters, J.C.; Ranheim, E.A.; Huebner, S.M.; Bristol, J.; Delecluse, H.J.; Kenney, S.C. Lmp1 and lmp2a collaborate to promote epstein-barr virus (ebv)-induced b cell lymphomas in a cord blood-humanized mouse model but are not essential. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Minamitani, T.; Ma, Y.; Zhou, H.; Kida, H.; Tsai, C.Y.; Obana, M.; Okuzaki, D.; Fujio, Y.; Kumanogoh, A.; Zhao, B.; et al. Mouse model of epstein-barr virus lmp1- and lmp2a-driven germinal center b-cell lymphoproliferative disease. Proc. Natl. Acad. Sci. USA 2017, 114, 4751–4756. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrzalikova, K.; Sunmonu, T.; Reynolds, G.; Murray, P. Contribution of Epstein–Barr Virus Latent Proteins to the Pathogenesis of Classical Hodgkin Lymphoma. Pathogens 2018, 7, 59. https://doi.org/10.3390/pathogens7030059
Vrzalikova K, Sunmonu T, Reynolds G, Murray P. Contribution of Epstein–Barr Virus Latent Proteins to the Pathogenesis of Classical Hodgkin Lymphoma. Pathogens. 2018; 7(3):59. https://doi.org/10.3390/pathogens7030059
Chicago/Turabian StyleVrzalikova, Katerina, Taofik Sunmonu, Gary Reynolds, and Paul Murray. 2018. "Contribution of Epstein–Barr Virus Latent Proteins to the Pathogenesis of Classical Hodgkin Lymphoma" Pathogens 7, no. 3: 59. https://doi.org/10.3390/pathogens7030059
APA StyleVrzalikova, K., Sunmonu, T., Reynolds, G., & Murray, P. (2018). Contribution of Epstein–Barr Virus Latent Proteins to the Pathogenesis of Classical Hodgkin Lymphoma. Pathogens, 7(3), 59. https://doi.org/10.3390/pathogens7030059