Navigating the Host Cell Response during Entry into Sites of Latent Cytomegalovirus Infection


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Clinical Manifestations of Human Cytomegalovirus
1.2. HCMV Latency
2. HCMV Entry
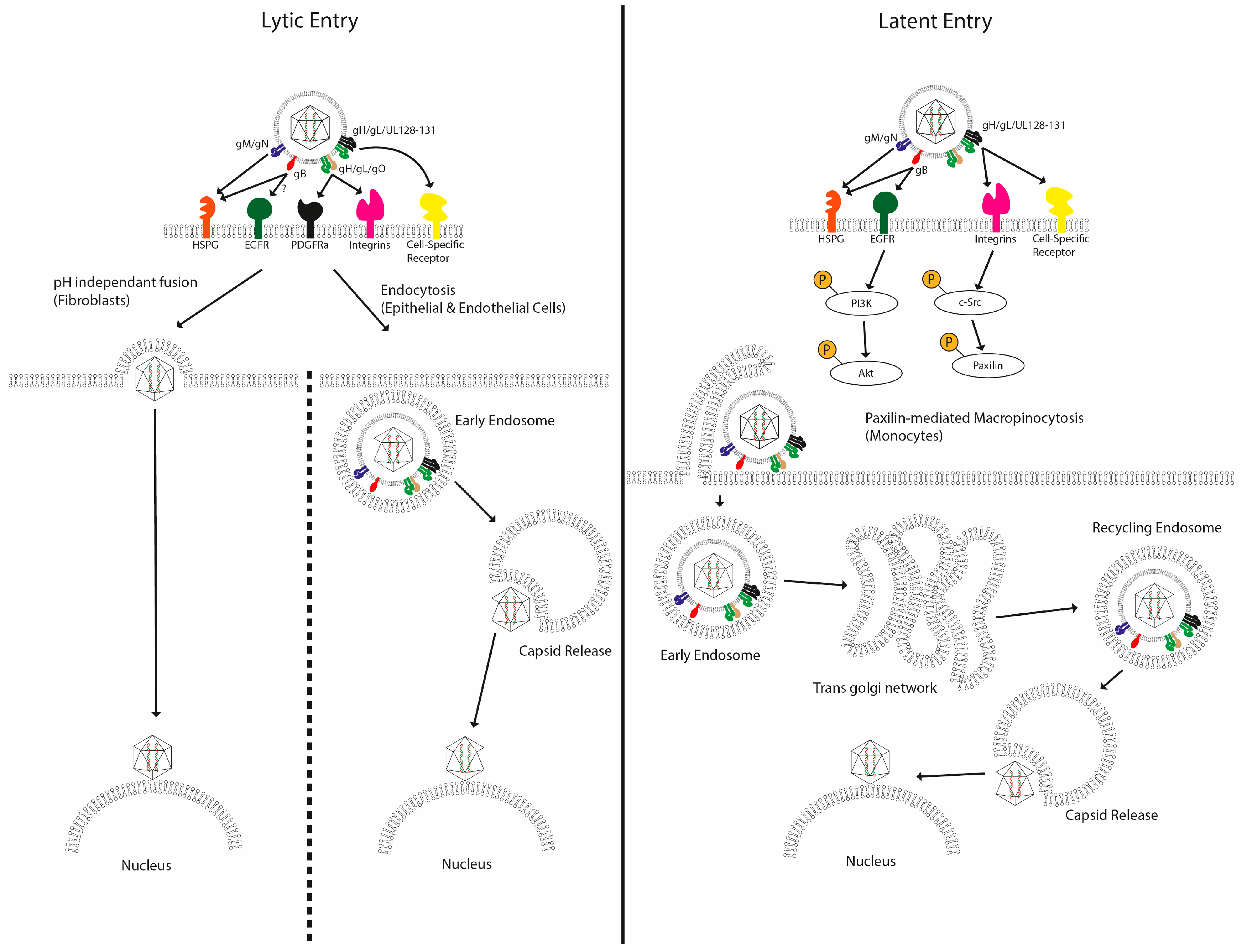
2.1. HCMV Entry into Permissive Cells
2.2. HCMV Entry into Non-Permissive Cells
3. Host Defences in Response to HCMV Infection
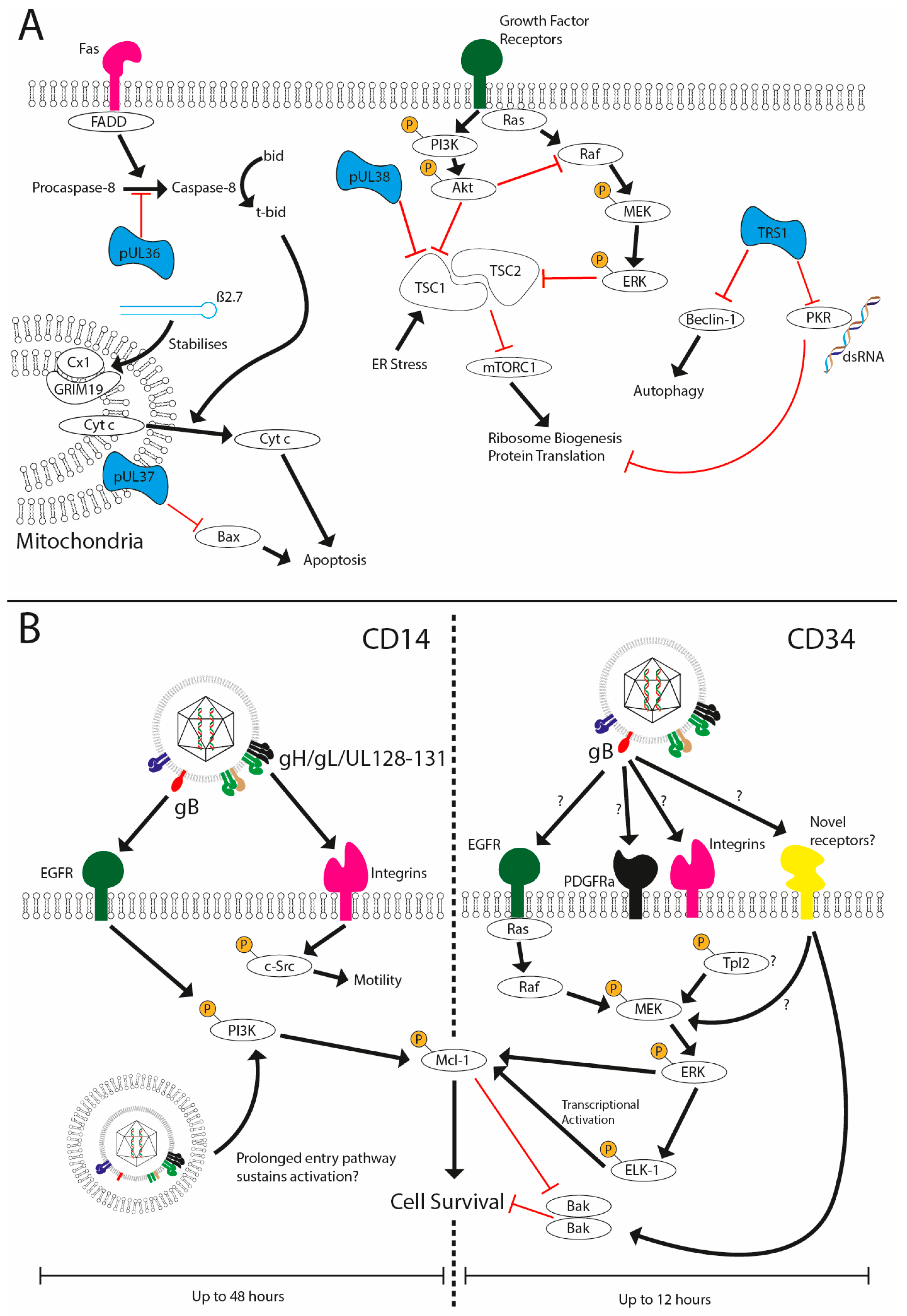
3.1. Evasion of Host Cell Apoptosis
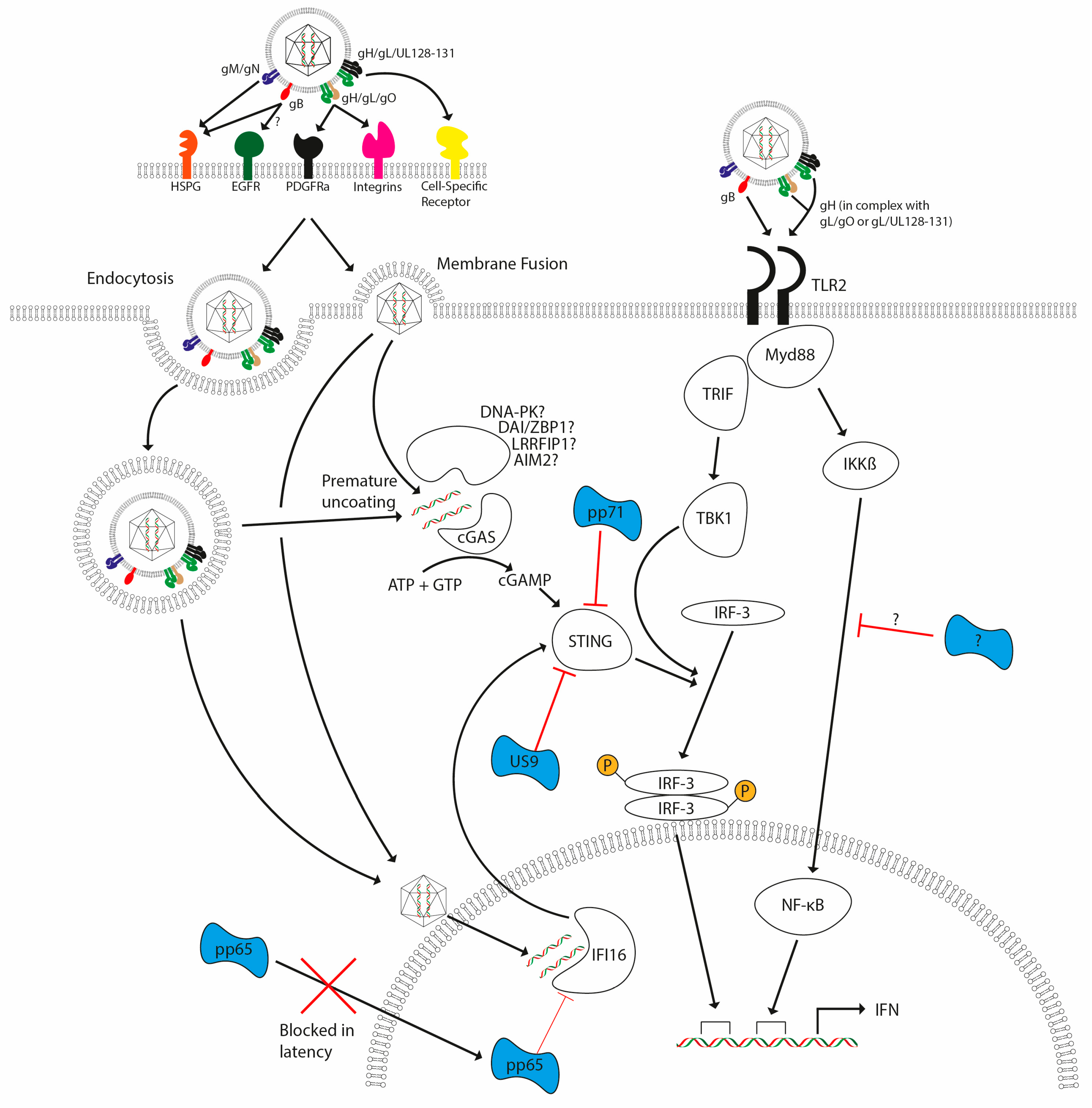
3.2. Innate Immune Mechanisms for Detection of CMV
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Randow, F.; MacMicking, J.D.; James, L.C. Cellular Self-Defense: How Cell-Autonomous Immunity Protects Against Pathogens. Science 2013, 340, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Weinrauch, Y.; Zychlinsky, A. The induction of apoptosis by bacterial pathogens. Annu. Rev. Microbiol. 1999, 53, 155–187. [Google Scholar] [CrossRef] [PubMed]
- Uyar, Y.; Balci, A.; Akcali, A.; Cabar, C. Prevalence of rubella and cytomegalovirus antibodies among pregnant women in northern Turkey. New Microbiol. 2008, 31, 451–455. [Google Scholar] [PubMed]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Deayton, J.R.; Wilson, P.; Sabin, C.A.; Davey, C.C.; Johnson, M.A.; Emery, V.C.; Griffiths, P.D. Changes in the natural history of cytomegalovirus retinitis following the introduction of highly active antiretroviral therapy. AIDS 2000, 14, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Razonable, R.R. Cytomegalovirus Infections in Solid Organ Transplantation: A Review. Infect. Chemother. 2013, 45, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.; Hakki, M.; Boeckh, M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol. Oncol. Clin. N. Am. 2011, 25, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D. Burden of disease associated with human cytomegalovirus and prospects for elimination by universal immunisation. Lancet Infect. Dis. 2012, 12, 790–798. [Google Scholar] [CrossRef]
- Dahle, A.J.; Fowler, K.B.; Wright, J.D.; Boppana, S.B.; Britt, W.J.; Pass, R.F. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J. Am. Acad. Audiol. 2000, 11, 283–290. [Google Scholar] [PubMed]
- Fowler, K.B. Congenital Cytomegalovirus Infection: Audiologic Outcome. Clin. Infect. Dis. 2013, 57, S182–S184. [Google Scholar] [CrossRef] [PubMed]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2009, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Sousa, H.; Boutolleau, D.; Ribeiro, J.; Teixeira, A.L.; Pinho Vaz, C.; Campilho, F.; Branca, R.; Campos, A., Jr.; Baldaque, I.; Medeiros, R. Cytomegalovirus infection in patients who underwent allogeneic hematopoietic stem cell transplantation in portugal: A five-year retrospective review. Biol. Blood Marrow Transplant. 2014, 20, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R.; National Vaccine Advisory Committee. Vaccine development to prevent cytomegalovirus disease: Report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar] [PubMed]
- Field, K.; Davies, M.E.; DeWitt, C.; Perry, H.C.; Liou, R.; Germershausen, J.; Karkas, J.D.; Ashton, W.T.; Johnston, D.B.; Tolman, R.L. 9-([2-Hydroxy-1-(Hydroxymethyl)Ethoxy]Methyl)Guanine: A Selective Inhibitor of Herpes Group Virus Replication. Proc. Natl. Acad. Sci. USA 1983, 80, 4139–4143. [Google Scholar] [CrossRef] [PubMed]
- Buhles, W.C.; Mastre, B.J.; Tinker, A.J.; Strand, V.; Koretz, S.H. Ganciclovir treatment of life-or sight-threatening cytomegalovirus infection: Experience in 314 immunocompromised patients. Rev. Infect. Dis. 1988, 10, S495–S506. [Google Scholar] [CrossRef] [PubMed]
- Salzberger, B.; Bowden, R.A.; Hackman, R.C.; Davis, C.; Boeckh, M. Neutropenia in allogeneic marrow transplant recipients receiving ganciclovir for prevention of cytomegalovirus disease: Risk factors and outcome. Blood 1997, 90, 2502–2508. [Google Scholar] [PubMed]
- Kim, E.S. Letermovir: First Global Approval. Drugs 2018, 78, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Siliciano, R.F. From reactivation of latent HIV-1 to elimination of the latent reservoir: The presence of multiple barriers to viral eradication. BioEssays 2013, 35, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Fauci, A.S. Latent reservoirs of HIV: Obstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA 1999, 96, 10958–10961. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Wills, M.; Sinclair, J. Human Cytomegalovirus Latency: Targeting Differences in the Latently Infected Cell with a View to Clearing Latent Infection. New J. Sci. 2014, 2014, 313761. [Google Scholar] [CrossRef]
- Krishna, B.A.; Lau, B.; Jackson, S.E.; Wills, M.R.; Sinclair, J.H.; Poole, E. Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci. Rep. 2016, 6, 24674. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Spiess, K.; Poole, E.L.; Lau, B.; Voigt, S.; Kledal, T.N.; Rosenkilde, M.M.; Sinclair, J.H. Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat. Commun. 2017, 8, 14321. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Tan, S.Y.L.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 2013, 340, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Sindre, H.; Tjøonnfjord, G.E.; Rollag, H.; Ranneberg-Nilsen, T.; Veiby, O.P.; Beck, S.; Degré, M.; Hestdal, K. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 1996, 88, 4526–4533. [Google Scholar] [PubMed]
- Taylor-Wiedeman, J.; Sissons, J.G.P.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Bevan, I.S.; Daw, R.A.; Day, P.J.; Ala, F.A.; Walker, M.R. Polymerase chain reaction for detection of human cytomegalovirus infection in a blood donor population. Br. J. Haematol. 1991, 78, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.P.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.; Sinclair, J. Regulation of human cytomegalovirus transcription in latency: Beyond the major immediate-early promoter. Viruses 2013, 5, 1395–1413. [Google Scholar] [CrossRef] [PubMed]
- Dupont, L.; Reeves, M.B. Cytomegalovirus latency and reactivation: Recent insights into an age old problem. Rev. Med. Virol. 2016, 26, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A Novel Viral Transcript with Homology to Human Interleukin-10 Is Expressed during Latent Human Cytomegalovirus Infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; St Jeor, S. Characterization of an Antisense Transcript Spanning the UL81-82 Locus of Human Cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Žugich, J.; Goodrum, F. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. USA 2017, 114, E10586–E10595. [Google Scholar] [CrossRef] [PubMed]
- Slobedman, B.; Mocarski, E.S. Quantitative Analysis of Latent Human Cytomegalovirus. J. Virol. 1999, 73, 4806–4812. [Google Scholar] [PubMed]
- Bolovan-Fritts, C.A.; Mocarski, E.S.; Wiedeman, J.A. Peripheral blood CD14(+) cells from healthy subjects carry a circular conformation of latent cytomegalovirus genome. Blood 1999, 93, 394–398. [Google Scholar] [PubMed]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res. 2011, 157, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Keeping it quiet: Chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 2013, 11, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Penkert, R.R.; Kalejta, R.F. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 2010, 84, 5594–5604. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways to Maintain Latent Infection. mBio 2017, 8, e01754-17. [Google Scholar] [CrossRef] [PubMed]
- Keyes, L.R.; Hargett, D.; Soland, M.; Bego, M.G.; Rossetto, C.C.; Almeida-Porada, G.; St. Jeor, S. HCMV Protein LUNA Is Required for Viral Reactivation from Latently Infected Primary CD14+ Cells. PLoS ONE 2012, 7, e52827. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Caviness, K.; Albright, E.R.; Lee, J.-H.; Gelbmann, C.B.; Rak, M.; Goodrum, F.; Kalejta, R.F. Long and short isoforms of the human cytomegalovirus UL138 protein silence IE1 transcription and promote latency. J. Virol. 2016, 90, 9483–9494. [Google Scholar] [CrossRef] [PubMed]
- Buehler, J.; Zeltzer, S.; Reitsma, J.; Petrucelli, A.; Umashankar, M.; Rak, M.; Zagallo, P.; Schroeder, J.; Terhune, S.; Goodrum, F. Opposing Regulation of the EGF Receptor: A Molecular Switch Controlling Cytomegalovirus Latency and Replication. PLoS Pathog. 2016, 12, e1005655. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Nowlin, D.M.; Cooper, N.R. Initiation of Human Cytomegalovirus Infection Requires Initial Interaction with Cell Surface Heparan Sulfate. Virology 1993, 193, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Kari, B.; Gehrz, R. A human cytomegalovirus glycoprotein complex designated gC-II is a major heparin-binding component of the envelope. J. Virol. 1992, 66, 1761–1764. [Google Scholar] [PubMed]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Chen, J.; Longnecker, R.; Jardetzky, T.S. The COMPLEXity in herpesvirus entry. Curr. Opin. Virol. 2007, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huong, S.M.; Chiu, M.L.; Raab-Traub, N.; Huang, E.S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, M.K.; Feire, A.L.; Compton, T. Epidermal Growth Factor Receptor Is Not Required for Human Cytomegalovirus Entry or Signaling. J. Virol. 2007, 81, 6241–6247. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, C.; Hochdorfer, D.; Lieber, D.; Subramanian, N.; Stöhr, D.; Laib Sampaio, K.; Sinzger, C. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PLoS Pathog. 2017, 13, e1006273. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Nepomuceno, R.R.; Nowlin, D.M. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 1992, 191, 387–395. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Compton, T. Human Cytomegalovirus Glycoprotein B Is Required for Virus Entry and Cell-to-Cell Spread but Not for Virion Attachment, Assembly, or Egress. J. Virol. 2009, 83, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 2008, 82, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Straschewski, S.; Patrone, M.; Walther, P.; Gallina, A.; Mertens, T.; Frascaroli, G. Protein pUL128 of Human Cytomegalovirus Is Necessary for Monocyte Infection and Blocking of Migration. J. Virol. 2011, 85, 5150–5158. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Chan, G.; Stevenson, E.V.; Gray, S.; Yurochko, A.D. Human Cytomegalovirus-Regulated Paxillin in Monocytes Links Cellular Pathogenic Motility to the Process of Viral Entry. J. Virol. 2011, 85, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Hetzenecker, S.; Helenius, A.; Krzyzaniak, M.A. HCMV Induces Macropinocytosis for Host Cell Entry in Fibroblasts. Traffic 2016, 17, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Compton, T. Inhibition of Inflammatory Interleukin-6 Activity via Extracellular Signal-Regulated Kinase-Mitogen-Activated Protein Kinase Signaling Antagonizes Human Cytomegalovirus Reactivation from Dendritic Cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Lathey, J.L.; Spector, S.A. Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J. Virol. 1991, 65, 6371–6375. [Google Scholar] [PubMed]
- Whitelaw, D.M. Observations on Human Monocyte Kinetics after Pulse Labeling. Cell Prolif. 1972, 5, 311–317. [Google Scholar] [CrossRef]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Gonczol, E.; Andrews, P.W.; Plotkin, S.A. Cytomegalovirus Infection of Human Teratocarcinoma Cells in Culture. J. Gen. Virol. 1985, 66, 509–515. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Murphy, E.A. A Myeloid Progenitor Cell Line Capable of Supporting Human Cytomegalovirus Latency and Reactivation, Resulting in Infectious Progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef] [PubMed]
- Albright, E.R.; Kalejta, R.F. Myeloblastic Cell Lines Mimic Some but Not All Aspects of Human Cytomegalovirus Experimental Latency Defined in Primary CD34+ Cell Populations. J. Virol. 2013, 87, 9802–9812. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. USA 2009, 106, 22369–22374. [Google Scholar] [CrossRef] [PubMed]
- Real, F.X.; Rettig, W.J.; Chesa, P.G.; Melamed, M.R.; Old, L.J.; Mendelsohn, J. Expression of epidermal growth factor receptor in human cultured cells and tissues: Relationship to cell lineage and stage of differentiation. Cancer Res. 1986, 46, 4726–4731. [Google Scholar] [PubMed]
- Kim, J.H.; Collins-McMillen, D.; Buehler, J.C.; Goodrum, F.D.; Yurochko, A.D. Human Cytomegalovirus Requires Epidermal Growth Factor Receptor Signaling to Enter and Initiate the Early Steps in the Establishment of Latency in CD34+ Human Progenitor Cells. J. Virol. 2017, 91, e01206-16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Collins-McMillen, D.; Caposio, P.; Yurochko, A.D. Viral binding-induced signaling drives a unique and extended intracellular trafficking pattern during infection of primary monocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 8819–8824. [Google Scholar] [CrossRef] [PubMed]
- Kew, V.G.; Wills, M.R.; Reeves, M.B. LPS promotes a monocyte phenotype permissive for human cytomegalovirus immediate-early gene expression upon infection but not reactivation from latency. Sci. Rep. 2017, 7, 810. [Google Scholar] [CrossRef] [PubMed]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef] [PubMed]
- Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.; Han, J.W.; Lutz, R.J.; Watanabe, S.; Cahir McFarland, E.D.; et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 1999, 96, 12536–12541. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human Cytomegalovirus UL38 Protein Blocks Apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The Human Cytomegalovirus Protein TRS1 Inhibits Autophagy via Its Interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human Cytomegalovirus Protein UL38 Inhibits Host Cell Stress Responses by Antagonizing the Tuberous Sclerosis Protein Complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Breidenstein, A.; Compton, T. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc. Natl. Acad. Sci. USA 2012, 109, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-Dependent Upregulation of Mcl-1 by Human Cytomegalovirus Is Mediated by Epidermal Growth Factor Receptor and Inhibits Apoptosis in Short-Lived Monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Kew, V.; Wills, M.; Reeves, M. HCMV activation of ERK-MAPK drives a multi-factorial response promoting the survival of infected myeloid progenitors. J. Mol. Biochem. 2017, 6, 13–25. [Google Scholar] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by AKT and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Iadevaia, V.; Liu, R.; Proud, C.G. MTORC1 signaling controls multiple steps in ribosome biogenesis. Semin. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human Cytomegalovirus Envelope Glycoproteins B and H Are Necessary for TLR2 Activation in Permissive Cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, M.E.; Khong, A.; Fleming, P.; Kuns, R.; Hertzog, P.J.; Frazer, I.H.; Andoniou, C.E.; Hill, G.R.; Degli-Esposti, M.A. The early monocytic response to cytomegalovirus infection is MyD88 dependent but occurs independently of common inflammatory cytokine signals. Eur. J. Immunol. 2014, 44, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Loewendorf, A.; De Trez, C.; Fulton, J.; Rhode, A.; Shumway, H.; Ha, S.; Patterson, G.; Pfeffer, K.; Nedospasov, S.A.; et al. Lymphotoxin-Mediated Crosstalk between B Cells and Splenic Stroma Promotes the Initial Type I Interferon Response to Cytomegalovirus. Cell Host Microbe 2008, 3, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Wang, Q.; Chodaczek, G.; Benedict, C.A. Lymphoid-Tissue Stromal Cells Coordinate Innate Defense to Cytomegalovirus. J. Virol. 2013, 87, 6201–6210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stetson, D.B.; Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012, 2012, e00047. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.E.; Ferguson, B.J.; Mazzon, M.; Fahy, A.S.; Krysztofinska, E.; Arribas-Bosacoma, R.; Pearl, L.H.; Ren, H.; Smith, G.L. A Mechanism for the Inhibition of DNA-PK-Mediated DNA Sensing by a Virus. PLoS Pathog. 2013, 9, e1003649. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W.J.; McDonald, B.; Takahashi, M.; Dhanwani, R.; Sharma, N.; Huang, J.; Pham, E.; Benedict, C.A.; Sharma, S. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J. Virol. 2016, 90, 7789–7797. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Su, S.; Gao, Y.Q.; Wang, P.P.; Huang, Z.F.; Hu, M.M.; Luo, W.W.; Li, S.; Luo, M.H.; Wang, Y.Y.; et al. Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Kalejta, R.F.; Bechtel, J.T.; Shenk, T. Human Cytomegalovirus pp71 Stimulates Cell Cycle Progression by Inducing the Proteasome-Dependent Degradation of the Retinoblastoma Family of Tumor Suppressors Human Cytomegalovirus pp71 Stimulates Cell Cycle Progression by Inducing the Proteasome-Depende. Mol. Cell. Biol. 2003, 23, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, V.R.; Alvarado, D.; Sali, T.; Rothenburg, S.; Fruh, K. Human Cytomegalovirus Induces the Interferon Response via the DNA Sensor ZBP1. J. Virol. 2010, 84, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choi, M.K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef] [PubMed]
- De Regge, N.; Van Opdenbosch, N.; Nauwynck, H.J.; Efstathiou, S.; Favoreel, H.W. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS ONE 2010, 5, e13076. [Google Scholar] [CrossRef] [PubMed]
- Dağ, F.; Dölken, L.; Holzki, J.; Drabig, A.; Weingärtner, A.; Schwerk, J.; Lienenklaus, S.; Conte, I.; Geffers, R.; Davenport, C.; et al. Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency. PLoS Pathog. 2014, 10, e1003962. [Google Scholar] [CrossRef] [PubMed]
- Kropp, K.A.; Robertson, K.A.; Sing, G.; Rodriguez-Martin, S.; Blanc, M.; Lacaze, P.; Hassim, M.F.B.N.; Khondoker, M.R.; Busche, A.; Dickinson, P.; et al. Reversible Inhibition of Murine Cytomegalovirus Replication by Gamma Interferon (IFN-γ) in Primary Macrophages Involves a Primed Type I IFN-Signaling Subnetwork for Full Establishment of an Immediate-Early Antiviral State. J. Virol. 2011, 85, 10286–10299. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, N.; Reuter, N.; Scherer, M.; Reichel, A.; Müller, R.; Stamminger, T. Contribution of the major ND10 proteins PML, hDaxx and sp100 to the regulation of human cytomegalovirus latency and lytic replication in the monocytic cell line THP-1. Viruses 2015, 7, 2884–2907. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.E.; Winship, D.; Snyder, J.M.; Child, S.J.; Geballe, A.P.; Stetson, D.B. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity 2016, 45, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 2007, 367, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 2009, 83, 6591–6598. [Google Scholar] [CrossRef] [PubMed]
- Schilling, E.M.; Otto, V.; Müller, R.; Reuter, N.; Stamminger, T. The human cytomegalovirus IE1 protein antagonizes ND10-mediated intrinsic immunity via the inhibition of PML de novo SUMOylation. J. Virol. 2017, 91, e02049-16. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; Kalejta, R.F. Nuclear localization of tegument-delivered pp71 in human cytomegalovirus-infected cells is facilitated by one or more factors present in terminally differentiated fibroblasts. J. Virol. 2010, 84, 9853–9863. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Sinclair, J.H. Knockdown of hDaxx in normally non-permissive undifferentiated cells does not permit human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 2007, 88, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, K.; Smith, M.S.; Malouli, D.; Mansouri, M.; Nelson, J.A.; Früh, K. BST2/tetherin enhances entry of human cytomegalovirus. PLoS Pathog. 2011, 7, e1002332. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Xuan, B.; Shan, J.; Pan, D.; Sun, Y.; Shan, Z.; Zhang, J.; Yu, D.; Li, B.; Qian, Z. Human Cytomegalovirus Exploits Interferon-Induced Transmembrane Proteins To Facilitate Morphogenesis of the Virion Assembly Compartment. J. Virol. 2015, 89, 3049–3061. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.L.; Huang, Y.; Kwok, H.Y.; Chen, M.; Chen, Z. Latent human cytomegalovirus enhances HIV-1 infection in CD34+ progenitor cells. Blood Adv. 2017, 1, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 2018, 72, 423.e25–438.e25. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murray, M.J.; Peters, N.E.; Reeves, M.B. Navigating the Host Cell Response during Entry into Sites of Latent Cytomegalovirus Infection. Pathogens 2018, 7, 30. https://doi.org/10.3390/pathogens7010030
Murray MJ, Peters NE, Reeves MB. Navigating the Host Cell Response during Entry into Sites of Latent Cytomegalovirus Infection. Pathogens. 2018; 7(1):30. https://doi.org/10.3390/pathogens7010030
Chicago/Turabian StyleMurray, Matthew J., Nicholas E. Peters, and Matthew B. Reeves. 2018. "Navigating the Host Cell Response during Entry into Sites of Latent Cytomegalovirus Infection" Pathogens 7, no. 1: 30. https://doi.org/10.3390/pathogens7010030
APA StyleMurray, M. J., Peters, N. E., & Reeves, M. B. (2018). Navigating the Host Cell Response during Entry into Sites of Latent Cytomegalovirus Infection. Pathogens, 7(1), 30. https://doi.org/10.3390/pathogens7010030