Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery


Abstract
1. Introduction
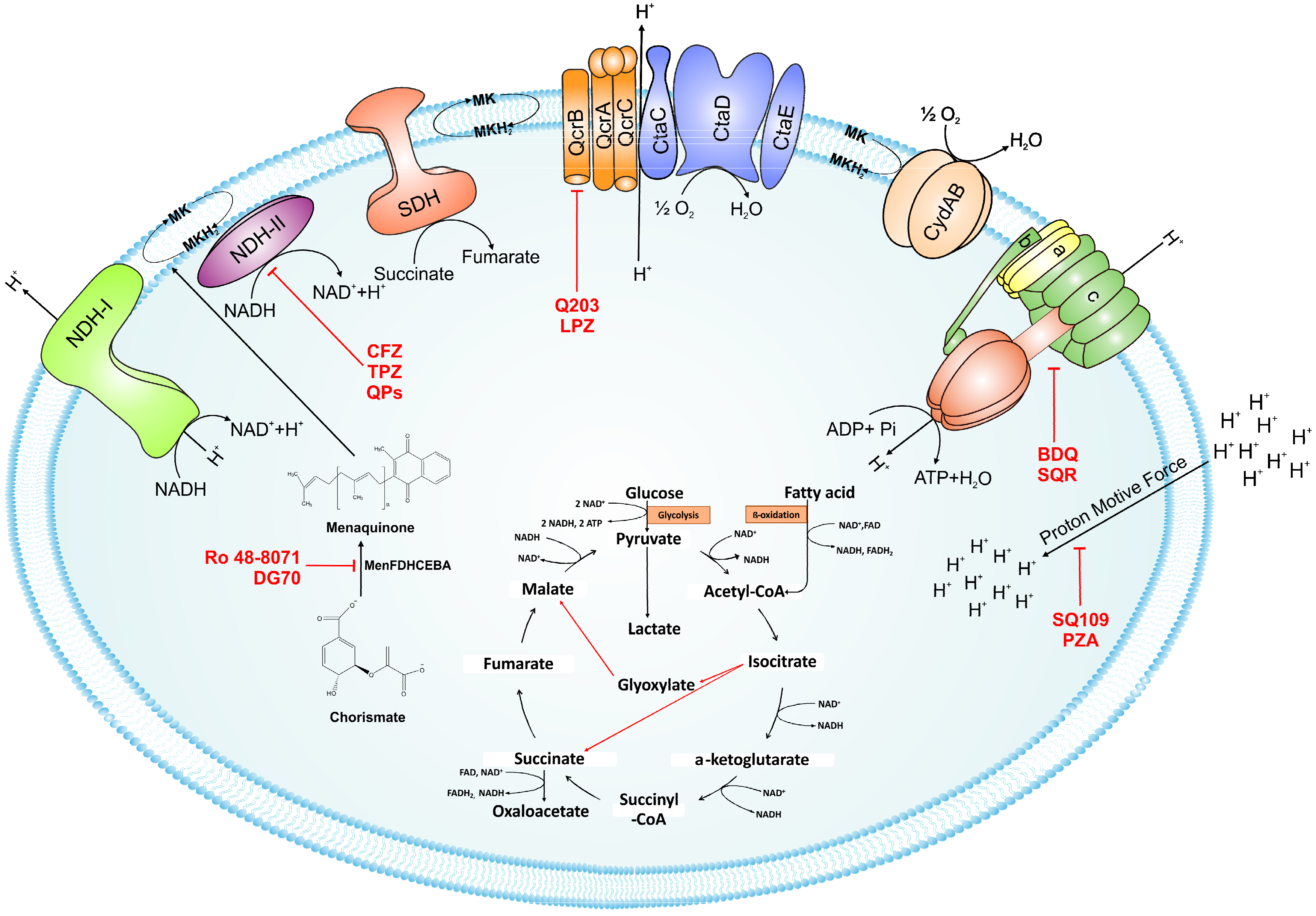
2. Feeding the Electrons into the Electron Transport Chain: Reduction of Menaquinone
2.1. NADH/Menaquinone Oxidoreductase
2.2. Inhibitors of NADH Dehydrogenase
2.3. Succinate Dehydrogenase
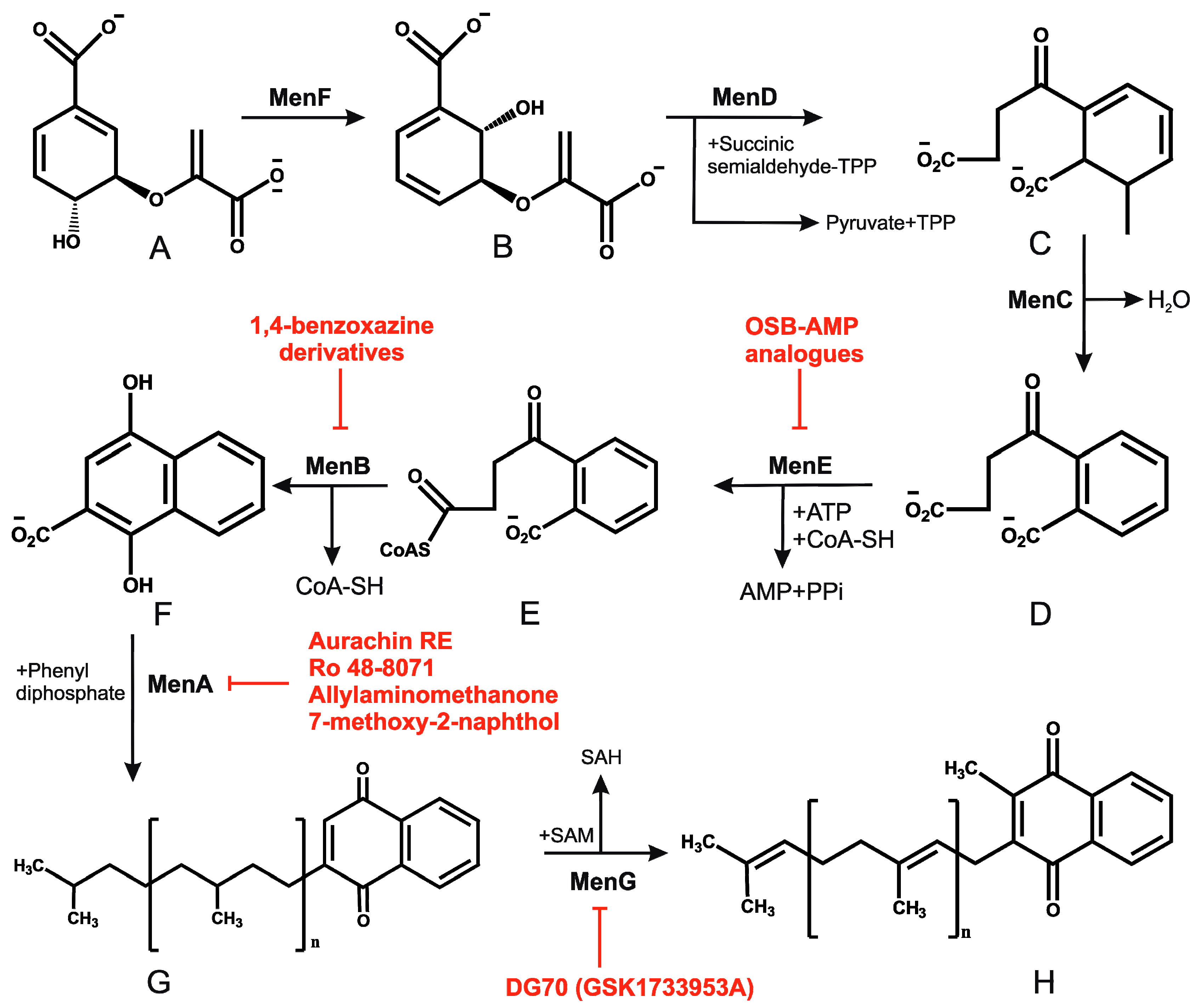
2.4. Menaquinone Biosynthesis and Inhibitors of Menaquinone Biosynthesis Pathway in Mtb
3. Oxidation of Menaquinone: A Tale of Two Terminal Oxidases
3.1. bc1-aa3 Pathway
3.2. Cytochrome bd-Oxidase
3.3. Supercomplex Inhibitors
4. ATP Synthase
4.1. Bedaquiline
4.2. Squaramides
5. Uncouplers of Proton Motive Force: Pyrazinamide and SQ109
6. Conclusions
Competing Financial Interests
Acknowledgments
Conflicts of Interest
Abbreviations
| 3NP | 3-Nitropropionate |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| BCG | Bacillus Calmette Guerin |
| BDQ | Bedaquiline |
| cfu | Colony forming units |
| CFZ | Clofazimine |
| CO2 | Carbon dioxide |
| cydKO | Cytochrome bd-oxidase knockout mutant |
| DCCD | N,N′-dicyclohexylcarbodiimide |
| DprE1 | Decaprenylphosphoryl-D-ribose oxidase |
| ETC | Electron transport chain |
| FADH2 | Reduced flavin adenine dinucleotide |
| FRD | Fumarate reductase |
| H+ | Proton |
| H2O | Water |
| HIV | Human immunodeficiency virus |
| HTS | High throughput screening |
| INH | Isoniazid |
| IP | Imidazo[1,2-a]pyridines |
| LPZ | Lansoprazole |
| LPZS | Lansoprazole sulphide |
| MDR | Multi drug resistant |
| MIC | Minimum inhibitory concentration |
| MK | Menaquinone |
| MKH2 | Menaquinol |
| Mtb | Mycobacterium tuberculosis |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NDH-1 | Type I NADH dehydrogenase |
| NDH-2 | Type II NADH dehydrogenase |
| NQR | Sodium-pumping NADH dehydrogenase |
| NTMs | Non-tuberculous mycobacteria |
| OCR | Oxygen consumption rate |
| OSB | O-succinyl-1-benzoate |
| PAB | Phenoxyalkylbenzimidazole |
| Pi | Inorganic phosphate |
| PMF | Proton motive force |
| POA | Pyrazinoic acid |
| PZA | Pyrazinamide |
| Q | Ubiquinone |
| RIF | Rifampicin |
| ROS | Reactive oxygen species |
| SAR | Structure–activity relationship |
| SDH | Succinate dehydrogenase |
| SNP | Single nucleotide polymorphism |
| TB | Tuberculosis |
| TCA cycle | Tricarboxylic acid cycle |
| TPZ | Trifluoperazine |
| TZ | Thioridazine |
| XDR | Extremely drug resistant |
References
- Boshoff, H.I.; Barry, C.E., 3rd. Tuberculosis—Metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 2005, 3, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Vander Ven, B.C.; Lee, W.; Abramovitch, R.B.; Kim, M.J.; Homolka, S.; Niemann, S.; Rohde, K.H. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 2010, 8, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, S.; Dröse, S.; Zickermann, V.; Brandt, U. The three families of respiratory NADH dehydrogenases. Results Probl. Cell Differ. 2008, 45, 185–222. [Google Scholar] [PubMed]
- Weinstein, E.A.; Yano, T.; Li, L.S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A.A.; Duncan, K.; Lonsdale, J.T.; Rubin, H. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. USA 2005, 102, 4548–4553. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.A.; Plikaytis, B.B.; Shinnick, T.M. Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 2002, 184, 4025–4032. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sohaskey, C.D.; Kana, B.D.; Dawes, S.; North, R.J.; Mizrahi, V.; Gennaro, M.L. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. USA 2005, 102, 15629–15634. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.; Alonso, S.; Rand, L.; Dick, T.; Pethe, K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11945–11950. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Iqbal, I.K.; Kumar, A. Imaging the NADH:NAD(+) Homeostasis for Understanding the Metabolic Response of Mycobacterium to Physiologically Relevant Stresses. Front. Cell. Infect. Microbiol. 2016, 6, 145. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, K.; Chen, B.; Miller, J.L.; Azogue, S.; Gurses, S.; Hsu, T.; Glickman, M.; Jacobs, W.R., Jr.; Porcelli, S.A.; Briken, V. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007, 3, e110. [Google Scholar] [CrossRef] [PubMed]
- Nantapong, N.; Otofuji, A.; Migita, C.T.; Adachi, O.; Toyama, H.; Matsushita, K. Electron transfer ability from NADH to menaquinone and from NADPH to oxygen of type II NADH dehydrogenase of Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 2005, 69, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Li, L.S.; Weinstein, E.; Teh, J.S.; Rubin, H. Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2). J. Biol. Chem. 2006, 281, 11456–11463. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yamashita, T.; Nakamaru-Ogiso, E.; Hashimoto, T.; Murai, M.; Igarashi, J.; Miyoshi, H.; Mori, N.; Matsuno-Yagi, A.; Yagi, T.; et al. Reaction mechanism of single subunit NADH-ubiquinone oxidoreductase (Ndi1) from Saccharomyces cerevisiae: Evidence for a ternary complex mechanism. J. Biol. Chem. 2011, 286, 9287–9297. [Google Scholar] [CrossRef] [PubMed]
- Sena, F.V.; Batista, A.P.; Catarino, T.; Brito, J.A.; Archer, M.; Viertler, M.; Madl, T.; Cabrita, E.J.; Pereira, M.M. Type-II NADH:quinone oxidoreductase from Staphylococcus aureus has two distinct binding sites and is rate limited by quinone reduction. Mol. Microbiol. 2015, 98, 272–288. [Google Scholar] [CrossRef] [PubMed]
- McAdam, R.A.; Quan, S.; Smith, D.A.; Bardarov, S.; Betts, J.C.; Cook, F.C.; Hooker, E.U.; Lewis, A.P.; Woollard, P.; Everett, M.J.; et al. Characterization of a mycobacterium tuberculosis h37rv transposon library reveals insertions in 351 orfs and mutants with altered virulence. Microbiology 2002, 148, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C.; et al. Transcriptional adaptation of mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Amewu, R.; Pacorel, B.; Nixon, G.L.; Gibbons, P.; Hong, W.D.; Leung, S.C.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, design and biological evaluation of bisaryl quinolones targeting plasmodium falciparum type ii nadh:Quinone oxidoreductase (pfndh2). J. Med. Chem. 2012, 55, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Kristiansen, J.E.; Viveiros, M.; Atouguia, J. Activity of phenothiazines against antibiotic-resistant mycobacterium tuberculosis: A review supporting further studies that may elucidate the potential use of thioridazine as anti-tuberculosis therapy. J. Antimicrob. Chemother. 2001, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Gadre, D.V.; Talwar, V.; Gupta, H.C.; Murthy, P.S. Effect of trifluoperazine, a potential drug for tuberculosis with psychotic disorders, on the growth of clinical isolates of drug resistant mycobacterium tuberculosis. Int. Clin. Psychopharmacol. 1998, 13, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Crowle, A.J.; Douvas, G.S.; May, M.H. Chlorpromazine: A drug potentially useful for treating mycobacterial infections. Chemotherapy 1992, 38, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Nadadhur, G.; Gangadharam, P.R. In-vitro and intracellular antimycobacterial activity of trifluoperazine. J. Antimicrob. Chemother. 1996, 37, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Katoch, V.M.; Saxena, N.; Shivannavar, C.T.; Sharma, V.D.; Katoch, K.; Sharma, R.K.; Murthy, P.S. Effect of trifluoperazine on in vitro atp synthesis by mycobacterium leprae. FEMS Immunol. Med. Microbiol. 1998, 20, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Van Soolingen, D.; Hernandez-Pando, R.; Orozco, H.; Aguilar, D.; Magis-Escurra, C.; Amaral, L.; van Ingen, J.; Boeree, M.J. The antipsychotic thioridazine shows promising therapeutic activity in a mouse model of multidrug-resistant tuberculosis. PLoS ONE 2010, 5, e12640. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.S.; Yano, T.; Rubin, H. Type ii nadh: Menaquinone oxidoreductase of mycobacterium tuberculosis. Infect. Disord. Drug Targets 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.D.; Gibbons, P.D.; Leung, S.C.; Amewu, R.; Stocks, P.A.; Stachulski, A.; Horta, P.; Cristiano, M.L.S.; Shone, A.E.; Moss, D.; et al. Rational design, synthesis, and biological evaluation of heterocyclic quinolones targeting the respiratory chain of mycobacterium tuberculosis. J. Med. Chem. 2017, 60, 3703–3726. [Google Scholar] [CrossRef] [PubMed]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Denneny, J.M.; Edward, D.W.; O'Sullivan, J.F.; Twomey, D.; Winder, F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Garrelts, J.C. Clofazimine: A review of its use in leprosy and mycobacterium avium complex infection. DICP Ann. Pharmacother. 1991, 25, 525–531. [Google Scholar] [CrossRef]
- O’Connor, R.; O’Sullivan, J.F.; O’Kennedy, R. The pharmacology, metabolism, and chemistry of clofazimine. Drug Metab. Rev. 1995, 27, 591–614. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; Nadadhur, G.; Daneluzzi, D.; O’Sullivan, J.F.; Gangadharam, P.R. Antituberculosis activities of clofazimine and its new analogs b4154 and b4157. Antimicrob. Agents Chemother. 1996, 40, 633–636. [Google Scholar] [PubMed]
- Yano, T.; Kassovska-Bratinova, S.; Teh, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 nadh: Quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ohnishi, T. The origin of cluster n2 of the energy-transducing nadh-quinone oxidoreductase: Comparisons of phylogenetically related enzymes. J. Bioenerg. Biomembr. 2001, 33, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.M.; Bandeiras, T.M.; Teixeira, M. New insights into type ii nad(p)h: Quinone oxidoreductases. Microbiol. Mol. Biol. Rev. 2004, 68, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; O’Sullivan, J.F.; Gangadharam, P.R. Antimycobacterial activities of riminophenazines. J. Antimicrob. Chemother. 1999, 43, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J. Turning the respiratory flexibility of mycobacterium tuberculosis against itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Paul, B.; Roy Choudhury, N.; Kedari, C.; Bandodkar, B.; Ugarkar, B.G. Quinolinyl pyrimidines: Potent inhibitors of ndh-2 as a novel class of anti-tb agents. ACS Med. Chem. Lett. 2012, 3, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Korkegian, A.; O’Malley, T.; Xia, Y.; Zhou, Y.; Carter, D.S.; Sunde, B.; Flint, L.; Thompson, D.; Ioerger, T.R.; Sacchettini, J.; et al. The 7-phenyl benzoxaborole series is active against Mycobacterium tuberculosis. Tuberculosis 2018, 108, 96–98. [Google Scholar] [CrossRef]
- Cecchini, G. Respiratory complex II: Role in cellular physiology and disease. Biochim. Biophys. Acta 2013, 1827, 541–542. [Google Scholar]
- Unden, G.; Bongaerts, J. Alternative respiratory pathways of Escherichia coli: Energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1997, 1320, 217–234. [Google Scholar] [CrossRef]
- Unden, G.; Schirawski, J. The oxygen-responsive transcriptional regulator FNR of Escherichia coli: The search for signals and reactions. Mol. Microbiol. 1997, 25, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kroger, A.; Biel, S.; Simon, J.; Gross, R.; Unden, G.; Lancaster, C.R. Fumarate respiration of wolinella succinogenes: Enzymology, energetics and coupling mechanism. Biochim. Biophys. Acta 2002, 1553, 23–38. [Google Scholar] [CrossRef]
- Eoh, H.; Rhee, K.Y. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, 6554–6559. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Zimmermann, M.; Goodwin, M.B.; Sauer, U.; Barry, C.E., 3rd; Boshoff, H.I. Fumarate reductase activity maintains an energized membrane in anaerobic mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002287. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.; Weinrick, B.; Vilcheze, C.; Berney, M.; Tufariello, J.; Cook, G.M.; Jacobs, W.R., Jr. Succinate dehydrogenase is the regulator of respiration in Mycobacterium tuberculosis. PLoS Pathog 2014, 10, e1004510. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, B.; Kruk, J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim. Biophys. Acta 2010, 1797, 1587–1605. [Google Scholar] [CrossRef] [PubMed]
- Knapczyk, J. Kinetics of sodium nitrite decomposition. Acta Poloniae Pharm. 1975, 32, 683–689. [Google Scholar] [PubMed]
- Azerad, R.; Cyrot-Pelletier, M.O. Structure and configuration of the polyprenoid side chain of dihydromenaquinones from Myco- and Corynebacteria. Biochimie 1973, 55, 591–603. [Google Scholar] [CrossRef]
- Dutton, R.J.; Wayman, A.; Wei, J.R.; Rubin, E.J.; Beckwith, J.; Boyd, D. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc. Natl. Acad. Sci. USA 2010, 107, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Toledo, J.C.; Patel, R.P.; Lancaster, J.R., Jr.; Steyn, A.J. Mycobacterium tuberculosis doss is a redox sensor and dost is a hypoxia sensor. Proc. Natl. Acad. Sci. USA 2007, 104, 11568–11573. [Google Scholar] [CrossRef] [PubMed]
- Honaker, R.W.; Dhiman, R.K.; Narayanasamy, P.; Crick, D.C.; Voskuil, M.I. Doss responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. J. Bacteriol. 2010, 192, 6447–6455. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.K.; Mahapatra, S.; Slayden, R.A.; Boyne, M.E.; Lenaerts, A.; Hinshaw, J.C.; Angala, S.K.; Chatterjee, D.; Biswas, K.; Narayanasamy, P.; et al. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol. 2009, 72, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, M.; Narayanasamy, P.; Biswas, K.; Dhiman, R.; Crick, D.C. Discovery of 1,4-dihydroxy-2-naphthoate [corrected] prenyltransferase inhibitors: New drug leads for multidrug-resistant gram-positive pathogens. J. Med. Chem. 2007, 50, 3973–3975. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, M.; Crick, D.C. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med. Chem. 2009, 5, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, W.; Tamura, T. A quinoline antibiotic from Rhodococcus erythropolis JCM 6824. J. Antibiot. 2008, 61, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Siricilla, S.; Wan, B.; Crick, D.C.; Lenaerts, A.J.; Franzblau, S.G.; Kurosu, M. Discovery of selective menaquinone biosynthesis inhibitors against mycobacterium tuberculosis. J. Med. Chem. 2012, 55, 3739–3755. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Larson, M.A.; Hinrichs, S.H.; Bartling, A.M.; Frandsen, J.; Narayanasamy, P. Discovery of bicyclic inhibitors against menaquinone biosynthesis. Future Med. Chem. 2016, 8, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, N.; Zhang, H.; Knudson, S.E.; Slayden, R.A.; Tonge, P.J. Synthesis and sar studies of 1,4-benzoxazine menb inhibitors: Novel antibacterial agents against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, X.; Liu, N.; Zhang, H.; Knudson, S.E.; Li, H.J.; Lai, C.T.; Simmerling, C.; Slayden, R.A.; Tonge, P.J. Coa adducts of 4-oxo-4-phenylbut-2-enoates: Inhibitors of menb from the m. Tuberculosis menaquinone biosynthesis pathway. ACS Med. Chem. Lett. 2011, 2, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, H.; Tonge, P.J.; Tan, D.S. Mechanism-based inhibitors of mene, an acyl-coa synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 2008, 18, 5963–5966. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, R.; Sharma, I.; Li, X.; Kumar, G.; Swaminathan, S.; Tonge, P.J.; Tan, D.S. Stable analogues of OSB-AMP: Potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone biosynthesis. Chembiochem Eur. J. Chem. Biol. 2012, 13, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Matarlo, J.S.; Evans, C.E.; Sharma, I.; Lavaud, L.J.; Ngo, S.C.; Shek, R.; Rajashankar, K.R.; French, J.B.; Tan, D.S.; Tonge, P.J. Mechanism of mene inhibition by acyl-adenylate analogues and discovery of novel antibacterial agents. Biochemistry 2015, 54, 6514–6524. [Google Scholar] [CrossRef] [PubMed]
- Sukheja, P.; Kumar, P.; Mittal, N.; Li, S.G.; Singleton, E.; Russo, R.; Perryman, A.L.; Shrestha, R.; Awasthi, D.; Husain, S.; et al. A Novel Small-Molecule Inhibitor of the Mycobacterium tuberculosis Demethylmenaquinone Methyltransferase MenG Is Bactericidal to Both Growing and Nutritionally Deprived Persister Cells. mBio 2017, 8, e02022-16. [Google Scholar] [CrossRef] [PubMed]
- Kana, B.D.; Weinstein, E.A.; Avarbock, D.; Dawes, S.S.; Rubin, H.; Mizrahi, V. Characterization of the cydab-encoded cytochrome bd oxidase from mycobacterium smegmatis. J. Bacteriol. 2001, 183, 7076–7086. [Google Scholar] [CrossRef] [PubMed]
- Matsoso, L.G.; Kana, B.D.; Crellin, P.K.; Lea-Smith, D.J.; Pelosi, A.; Powell, D.; Dawes, S.S.; Rubin, H.; Coppel, R.L.; Mizrahi, V. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 2005, 187, 6300–6308. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, R.; Hill, S.; Poole, R.K. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: Implications for regulation of activity in vivo by oxygen inhibition. Microbiology 1996, 142 Pt 4, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y. Proton translocation by the cytochrome bc1 complexes of phototrophic bacteria: Introducing the activated Q-cycle. Photochem. Photobiol. Sci. 2007, 6, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Niebisch, A.; Bott, M. Molecular analysis of the cytochrome bc1-aa3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch. Microbiol. 2001, 175, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.A.; Guergova-Kuras, M.; Huang, L.S.; Crofts, A.R. Structure and function of cytochrome bc complexes. Annu. Rev. Biochem. 2000, 69, 1005–1075. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Hards, K.; Vilcheze, C.; Hartman, T.; Berney, M. Energetics of Respiration and Oxidative Phosphorylation in Mycobacteria. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Niebisch, A.; Bott, M. Purification of a cytochrome bc-aa3 supercomplex with quinol oxidase activity from Corynebacterium glutamicum. Identification of a fourth subunity of cytochrome aa3 oxidase and mutational analysis of diheme cytochrome c1. J. Biol. Chem. 2003, 278, 4339–4346. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.; Shibata, T.; Mine, T.; Miyahara, R.; Torigoe, T.; Noguchi, S.; Matsushita, K.; Sone, N. Cytochrome c oxidase contains an extra charged amino acid cluster in a new type of respiratory chain in the amino-acid-producing Gram-positive bacterium Corynebacterium glutamicum. Microbiology 2001, 147, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Sone, N.; Fukuda, M.; Katayama, S.; Jyoudai, A.; Syugyou, M.; Noguchi, S.; Sakamoto, J. QcrCAB operon of a nocardia-form actinomycete Rhodococcus rhodochrous encodes cytochrome reductase complex with diheme cytochrome cc subunit. Biochim. Biophys. Acta 2003, 1557, 125–131. [Google Scholar] [CrossRef]
- Sone, N.; Nagata, K.; Kojima, H.; Tajima, J.; Kodera, Y.; Kanamaru, T.; Noguchi, S.; Sakamoto, J. A novel hydrophobic diheme c-type cytochrome. Purification from Corynebacterium glutamicum and analysis of the QcrCBA operon encoding three subunit proteins of a putative cytochrome reductase complex. Biochim. Biophys. Acta 2001, 1503, 279–290. [Google Scholar] [CrossRef]
- Megehee, J.A.; Hosler, J.P.; Lundrigan, M.D. Evidence for a cytochrome bcc-aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiology 2006, 152, 823–829. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.S.; Jang, J.; Ab Rahman, N.B.; Pethe, K.; Berry, E.A.; Huang, L.S. Isolation and Characterization of a Hybrid Respiratory Supercomplex Consisting of Mycobacterium tuberculosis Cytochrome bcc and Mycobacterium smegmatis Cytochrome aa3. J. Biol. Chem. 2015, 290, 14350–14360. [Google Scholar] [CrossRef] [PubMed]
- Goldman, B.S.; Gabbert, K.K.; Kranz, R.G. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J. Bacteriol. 1996, 178, 6348–6351. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.K.; Cook, G.M. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv. Microb. Physiol. 2000, 43, 165–224. [Google Scholar] [PubMed]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Borisov, V.B.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffre, A. Cytochrome bd oxidase and hydrogen peroxide resistance in mycobacterium tuberculosis. MBio 2013, 4, e01006-01013. [Google Scholar] [CrossRef] [PubMed]
- Giuffre, A.; Borisov, V.B.; Arese, M.; Sarti, P.; Forte, E. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim. Biophys. Acta 2014, 1837, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Konishi, K.; Anraku, Y. Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 1984, 259, 3375–3381. [Google Scholar] [PubMed]
- Roberts, G.; Vadrevu, I.S.; Madiraju, M.V.; Parish, T. Control of CydB and GltA1 expression by the SenX3 RegX3 two component regulatory system of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e21090. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Kumar, A. Virulence factor SenX3 is the oxygen-controlled replication switch of Mycobacterium tuberculosis. Antioxid. Redox Signal. 2015, 22, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; van Spanning, R.; Bald, D. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef] [PubMed]
- Boot, M.; Jim, K.K.; Liu, T.; Commandeur, S.; Lu, P.; Verboom, T.; Lill, H.; Bitter, W.; Bald, D. A fluorescence-based reporter for monitoring expression of mycobacterial cytochrome bd in response to antibacterials and during infection. Sci. Rep. 2017, 7, 10665. [Google Scholar] [CrossRef] [PubMed]
- Way, S.S.; Sallustio, S.; Magliozzo, R.S.; Goldberg, M.B. Impact of either elevated or decreased levels of cytochrome bd expression on Shigella flexneri virulence. J. Bacteriol. 1999, 181, 1229–1237. [Google Scholar] [PubMed]
- Endley, S.; McMurray, D.; Ficht, T.A. Interruption of the cydB locus in Brucella abortus attenuates intracellular survival and virulence in the mouse model of infection. J. Bacteriol. 2001, 183, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.K.; Barber, L.Z.; Wigley, P.; Muhammad, S.; Jones, M.A.; Lovell, M.A.; Hulme, S.; Barrow, P.A. Contribution of proton-translocating proteins to the virulence of Salmonella enterica serovars Typhimurium, Gallinarum, and Dublin in chickens and mice. Infect. Immun. 2003, 71, 3392–3401. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Herrmann, J.; Zang, Y.; Grellier, P.; Prado, S.; Muller, R.; Nay, B. Synthesis and biological activities of the respiratory chain inhibitor aurachin D and new ring versus chain analogues. Beilstein J. Org. Chem. 2013, 9, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Von Jagow, G.; Ljungdahl, P.O.; Graf, P.; Ohnishi, T.; Trumpower, B.L. An inhibitor of mitochondrial respiration which binds to cytochrome b and displaces quinone from the iron-sulfur protein of the cytochrome bc1 complex. J. Biol. Chem. 1984, 259, 6318–6326. [Google Scholar] [PubMed]
- Thierbach, G.; Reichenbach, H. Myxothiazol, a new antibiotic interfering with respiration. Antimicrob. Agents Chemother. 1981, 19, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Von Jagow, G.; Gribble, G.W.; Trumpower, B.L. Mucidin and strobilurin A are identical and inhibit electron transfer in the cytochrome bc1 complex of the mitochondrial respiratory chain at the same site as myxothiazol. Biochemistry 1986, 25, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Kucera, I.; Hedbavny, R.; Dadak, V. Separate binding sites for antimycin and mucidin in the respiratory chain of the bacterium Paracoccus denitrificans and their occurrence in other denitrificans bacteria. Biochem. J. 1988, 252, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Kemmer, T.; Hofle, G.; Reichenbach, H. Stigmatellin, a new antibiotic from Stigmatella aurantiaca (Myxobacterales). I. Production, physico-chemical and biological properties. J. Antibiot. 1984, 37, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Thierbach, G.; Kunze, B.; Reichenbach, H.; Höfle, G. The mode of action of stigmatellin, a new inhibitor of the cytochrome b-c1 segment of the respiratory chain. Biochim. Biophys. Acta 1984, 765, 227–235. [Google Scholar] [CrossRef]
- Mak, P.A.; Rao, S.P.; Ping Tan, M.; Lin, X.; Chyba, J.; Tay, J.; Ng, S.H.; Tan, B.H.; Cherian, J.; Duraiswamy, J.; et al. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Cox, J.A.; Spivey, V.L.; Loman, N.J.; Pallen, M.J.; Constantinidou, C.; Fernandez, R.; Alemparte, C.; Remuinan, M.J.; Barros, D.; et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 2012, 7, e52951. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Faaleolea, E.R.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Laughon, B.E.; Maddry, J.A.; Mehta, A.; Rasmussen, L.; Reynolds, R.C.; et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, N.S.; Alling, T.; Bailey, M.A.; Files, M.; Early, J.V.; Ollinger, J.; Ovechkina, Y.; Masquelin, T.; Desai, P.V.; Cramer, J.W.; et al. Identification of Phenoxyalkylbenzimidazoles with Antitubercular Activity. J. Med. Chem. 2015, 58, 7273–7285. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, N.S.; Berube, B.J.; Shetye, G.; Chettiar, S.; O'Malley, T.; Manning, A.; Flint, L.; Awasthi, D.; Ioerger, T.R.; Sacchettini, J.; et al. Improved Phenoxyalkylbenzimidazoles with Activity against Mycobacterium tuberculosis Appear to Target QcrB. ACS Infect. Dis. 2017, 3, 898–916. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Xu, Y.J.; Tu, H. Regiospecific synthesis of 3-substituted imidazo[1,2-a]pyridines, imidazo[1,2-a]pyrimidines, and imidazo[1,2-c]pyrimidine. J. Org. Chem. 2003, 68, 4935–4937. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Seeger, N.; Miller, P.A.; Oliver, A.G.; Boshoff, H.I.; Cho, S.; Mulugeta, S.; Anderson, J.R.; Franzblau, S.G.; Miller, M.J. Arrival of Imidazo[2,1-b]thiazole-5-carboxamides: Potent Anti-tuberculosis Agents That Target QcrB. ACS Infect. Dis. 2016, 2, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Cheng, Y.; Cho, S.; Cramer, J.W.; Godfrey, A.; Masquelin, T.; Franzblau, S.G.; Miller, M.J.; Schorey, J. Imidazo[1,2-a]Pyridine-3-Carboxamides Are Active Antimicrobial Agents against Mycobacterium avium Infection In Vivo. Antimicrob. Agents Chemother. 2016, 60, 5018–5022. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.H.; Smith, P.W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorgan. Med. Chem. 2015, 23, 5087–5097. [Google Scholar] [CrossRef] [PubMed]
- Van der Westhuyzen, R.; Winks, S.; Wilson, C.R.; Boyle, G.A.; Gessner, R.K.; Soares de Melo, C.; Taylor, D.; de Kock, C.; Njoroge, M.; Brunschwig, C.; et al. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: A Novel Antimycobacterial Class Targeting Mycobacterial Respiration. J. Med. Chem. 2015, 58, 9371–9381. [Google Scholar] [CrossRef] [PubMed]
- Rybniker, J.; Vocat, A.; Sala, C.; Busso, P.; Pojer, F.; Benjak, A.; Cole, S.T. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 2015, 6, 7659. [Google Scholar] [CrossRef] [PubMed]
- Esser, L.; Elberry, M.; Zhou, F.; Yu, C.A.; Yu, L.; Xia, D. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. J. Biol. Chem. 2008, 283, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Mdanda, S.; Baijnath, S.; Shobo, A.; Singh, S.D.; Maguire, G.E.M.; Kruger, H.G.; Arvidsson, P.I.; Naicker, T.; Govender, T. Lansoprazole-sulfide, pharmacokinetics of this promising anti-tuberculous agent. Biomed. Chromatogr. BMC 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Yates, T.A.; Tomlinson, L.A.; Bhaskaran, K.; Langan, S.; Thomas, S.; Smeeth, L.; Douglas, I.J. Lansoprazole use and tuberculosis incidence in the United Kingdom Clinical Practice Research Datalink: A population based cohort. PLoS Med. 2017, 14, e1002457. [Google Scholar] [CrossRef] [PubMed]
- Kalia, N.P.; Hasenoehrl, E.J.; Ab Rahman, N.B.; Koh, V.H.; Ang, M.L.T.; Sajorda, D.R.; Hards, K.; Gruber, G.; Alonso, S.; Cook, G.M.; et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl. Acad. Sci. USA 2017, 114, 7426–7431. [Google Scholar] [CrossRef] [PubMed]
- Moosa, A.; Lamprecht, D.A.; Arora, K.; Barry, C.E., 3rd; Boshoff, H.I.M.; Ioerger, T.R.; Steyn, A.J.C.; Mizrahi, V.; Warner, D.F. Susceptibility of Mycobacterium tuberculosis Cytochrome bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrob. Agents Chemother. 2017, 61, e01338-17. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, P.E.; Von Groll, A.; Martin, A.; Palomino, J.C. Efflux as a mechanism for drug resistance in Mycobacterium tuberculosis. FEMS Immunol. Med. Microbiol. 2011, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Kim, R.; Woo, M.; Jeong, J.; Park, D.E.; Kim, G.; Delorme, V. Efflux Attenuates the Antibacterial Activity of Q203 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Laubinger, W.; Dimroth, P. Characterization of the ATP synthase of Propionigenium modestum as a primary sodium pump. Biochemistry 1988, 27, 7531–7537. [Google Scholar] [CrossRef] [PubMed]
- Reidlinger, J.; Muller, V. Purification of ATP synthase from Acetobacterium woodii and identification as a Na(+)-translocating F1F0-type enzyme. Eur. J. Biochem. 1994, 223, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.L.; Fillingame, R.H. Stoichiometry of subunits in the H+-ATPase complex of Escherichia coli. J. Biol. Chem. 1982, 257, 2009–2015. [Google Scholar] [PubMed]
- Stock, D.; Leslie, A.G.; Walker, J.E. Molecular architecture of the rotary motor in ATP synthase. Science 1999, 286, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Sambongi, Y.; Iko, Y.; Tanabe, M.; Omote, H.; Iwamoto-Kihara, A.; Ueda, I.; Yanagida, T.; Wada, Y.; Futai, M. Mechanical rotation of the c subunit oligomer in ATP synthase (F0F1): Direct observation. Science 1999, 286, 1722–1724. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ueno, H.; Mitome, N.; Suzuki, J.; Yoshida, M. F(0) of ATP synthase is a rotary proton channel. Obligatory coupling of proton translocation with rotation of c-subunit ring. J. Biol. Chem. 2002, 277, 13281–13285. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Farhana, A.; Guidry, L.; Saini, V.; Hondalus, M.; Steyn, A.J. Redox homeostasis in mycobacteria: The key to tuberculosis control? Expert Rev. Mol. Med. 2011, 13, e39. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Singh, N.; Trivedi, A.; Kansal, P.; Gupta, P.; Kumar, A. The mechanism of redox sensing in Mycobacterium tuberculosis. Free Radic. Biol. Med. 2012, 53, 1625–1641. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Singh, N.; Bhat, S.A.; Gupta, P.; Kumar, A. Redox biology of tuberculosis pathogenesis. Adv. Microb. Physiol. 2012, 60, 263–324. [Google Scholar] [PubMed]
- Tran, S.L.; Cook, G.M. The F1Fo-ATP synthase of Mycobacterium smegmatis is essential for growth. J. Bacteriol. 2005, 187, 5023–5028. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Post, F.A.; Bekker, L.G.; Walther, G.B.; Wainwright, H.C.; Manganelli, R.; Chan, W.T.; Tsenova, L.; Gold, B.; Smith, I.; et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. USA 2003, 100, 14321–14326. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Rao, S.P.S.; Pethe, K.; Dick, T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology 2010, 156, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, M.; Tam, A.; Steinberg, D. Differential gene expression profiling of Streptococcus mutans cultured under biofilm and planktonic conditions. Microbiology 2007, 153, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Mavi, P.S.; Bhatt, D.; Kumar, A. Thiol reductive stress induces cellulose-anchored biofilm formation in Mycobacterium tuberculosis. Nat. Commun. 2016, 7, 11392. [Google Scholar] [CrossRef] [PubMed]
- Maglica, Z.; Ozdemir, E.; McKinney, J.D. Single-cell tracking reveals antibiotic-induced changes in mycobacterial energy metabolism. mBio 2015, 6, e02236-14. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Haouz, A.; Saul, F.A.; Miras, I.; Rosenkrands, I.; Alzari, P.M.; Cole, S.T. Genome-wide regulon and crystal structure of BlaI (Rv1846c) from Mycobacterium tuberculosis. Mol. Microbiol. 2009, 71, 1102–1116. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta 2014, 1837, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Kashket, E.R. Effects of aerobiosis and nitrogen source on the proton motive force in growing Escherichia coli and Klebsiella pneumoniae cells. J. Bacteriol. 1981, 146, 377–384. [Google Scholar] [PubMed]
- Haagsma, A.C.; Driessen, N.N.; Hahn, M.M.; Lill, H.; Bald, D. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 2010, 313, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Kalra, V.K.; Lee, S.H.; Bogin, E.; Brodie, A.F. Energy-transducing membrane-bound coupling factor-ATPase from Mycobacterium phlei. I. Purification, homogeneity, and properties. J. Biol. Chem. 1975, 250, 6541–6548. [Google Scholar] [PubMed]
- Von Ballmoos, C.; Wiedenmann, A.; Dimroth, P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu. Rev. Biochem. 2009, 78, 649–672. [Google Scholar] [CrossRef] [PubMed]
- Ragunathan, P.; Sielaff, H.; Sundararaman, L.; Biukovic, G.; Subramanian Manimekalai, M.S.; Singh, D.; Kundu, S.; Wohland, T.; Frasch, W.; Dick, T.; et al. The uniqueness of subunit alpha of mycobacterial F-ATP synthases: An evolutionary variant for niche adaptation. J. Biol. Chem. 2017, 292, 11262–11279. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Biukovic, G.; Manimekalai, M.S.; Lim, J.; Rao, S.P.; Gruber, G. Solution structure of subunit gamma (gamma(1-204)) of the Mycobacterium tuberculosis F-ATP synthase and the unique loop of gamma(165-178), representing a novel TB drug target. J. Bioenerg. Biomembr. 2013, 45, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Hotra, A.; Suter, M.; Biukovic, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Gruber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase gamma subunit sheds light on its inhibitory role in ATP hydrolysis-driven H(+) pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Matthies, D.; Preiss, L.; Klyszejko, A.L.; Muller, D.J.; Cook, G.M.; Vonck, J.; Meier, T. The c13 ring from a thermoalkaliphilic ATP synthase reveals an extended diameter due to a special structural region. J. Mol. Biol. 2009, 388, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.; Morgner, N.; Matthies, D.; Pogoryelov, D.; Keis, S.; Cook, G.M.; Dimroth, P.; Brutschy, B. A tridecameric c ring of the adenosine triphosphate (ATP) synthase from the thermoalkaliphilic Bacillus sp. strain TA2.A1 facilitates ATP synthesis at low electrochemical proton potential. Mol. Microbiol. 2007, 65, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.B.; Liu, J.; Fujisawa, M.; Krulwich, T.A. F1F0-ATP synthases of alkaliphilic bacteria: Lessons from their adaptations. Biochim. Biophys. Acta 2010, 1797, 1362–1377. [Google Scholar] [CrossRef] [PubMed]
- Preiss, L.; Langer, J.D.; Yildiz, O.; Eckhardt-Strelau, L.; Guillemont, J.E.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef] [PubMed]
- Beechey, R.B.; Holloway, C.T.; Knight, I.G.; Roberton, A.M. Dicyclohexylcarbodiimide—An inhibitor of oxidative phosphorylation. Biochem. Biophys. Res. Commun. 1966, 23, 75–80. [Google Scholar] [CrossRef]
- Matsuno-Yagi, A.; Hatefi, Y. Studies on the mechanism of oxidative phosphorylation. Different effects of F0 inhibitors on unisite and multisite ATP hydrolysis by bovine submitochondrial particles. J. Biol. Chem. 1993, 268, 1539–1545. [Google Scholar] [PubMed]
- Hong, S.; Pedersen, P.L. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol. Mol. Biol. Rev. MMBR 2008, 72, 590–641. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Gohlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Janssen Pharmaceuticals, I. Quinoline Derivatives and Their Use as Mycobacterial Inhibitors. Patent EP1527050 A1, 2005. [Google Scholar]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; Van den Wyngaert, I.; Vergauwen, K.; Gohlmann, H.W.; Willebrords, R.; Poncelet, A.; Guillemont, J.; et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef] [PubMed]
- Veziris, N.; Ibrahim, M.; Lounis, N.; Andries, K.; Jarlier, V. Sterilizing activity of second-line regimens containing TMC207 in a murine model of tuberculosis. PLoS ONE 2011, 6, e17556. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Li, S.Y.; Peloquin, C.A.; Taylor, D.; Williams, K.N.; Andries, K.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 5485–5492. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Minkowski, A.; Amoabeng, O.; Peloquin, C.A.; Taylor, D.; Andries, K.; Wallis, R.S.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3114–3120. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Pym, A.; Grobusch, M.P.; de los Rios, J.M.; Gotuzzo, E.; Vasilyeva, I.; Leimane, V.; Andries, K.; Bakare, N.; De Marez, T.; et al. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N. Engl. J. Med. 2014, 371, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G.; et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, A.C.; Abdillahi-Ibrahim, R.; Wagner, M.J.; Krab, K.; Vergauwen, K.; Guillemont, J.; Andries, K.; Lill, H.; Koul, A.; Bald, D. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 2009, 53, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Vesenbeckh, S.; Schonfeld, N.; Roth, A.; Bettermann, G.; Krieger, D.; Bauer, T.T.; Russmann, H.; Mauch, H. Bedaquiline as a potential agent in the treatment of Mycobacterium abscessus infections. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed]
- Vesenbeckh, S.; Schonfeld, N.; Krieger, D.; Bettermann, G.; Bauer, T.T.; Russmann, H.; Mauch, H. Bedaquiline as a potential agent in the treatment of M. intracellulare and M. avium infections. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef]
- Dupont, C.; Viljoen, A.; Thomas, S.; Roquet-Baneres, F.; Herrmann, J.L.; Pethe, K.; Kremer, L. Bedaquiline Inhibits the ATP Synthase in Mycobacterium abscessus and Is Effective in Infected Zebrafish. Antimicrob. Agents Chemother. 2017, 61, e01225-17. [Google Scholar] [CrossRef] [PubMed]
- Gelber, R.; Andries, K.; Paredes, R.M.; Andaya, C.E.; Burgos, J. The diarylquinoline R207910 is bactericidal against Mycobacterium leprae in mice at low dose and administered intermittently. Antimicrob. Agents Chemother. 2009, 53, 3989–3991. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lounis, N.; Gevers, T.; Van den Berg, J.; Vranckx, L.; Andries, K. ATP synthase inhibition of Mycobacterium avium is not bactericidal. Antimicrob. Agents Chemother. 2009, 53, 4927–4929. [Google Scholar] [CrossRef] [PubMed]
- Lerat, I.; Cambau, E.; Roth Dit Bettoni, R.; Gaillard, J.L.; Jarlier, V.; Truffot, C.; Veziris, N. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J. Infect. Dis. 2014, 209, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef] [PubMed]
- Lounis, N.; Veziris, N.; Chauffour, A.; Truffot-Pernot, C.; Andries, K.; Jarlier, V. Combinations of R207910 with drugs used to treat multidrug-resistant tuberculosis have the potential to shorten treatment duration. Antimicrob. Agents Chemother. 2006, 50, 3543–3547. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Andries, K.; Lounis, N.; Chauffour, A.; Truffot-Pernot, C.; Jarlier, V.; Veziris, N. Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; Einck, L.; Andries, K.; Nacy, C.A. In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 2010, 54, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Lechartier, B.; Hartkoorn, R.C.; Cole, S.T. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5790–5793. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Manina, G.; Mikusova, K.; Mollmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.M.; Murray, S.; Karlsson, M.O.; Dooley, K.E. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti-TB drug. J. Antimicrob. Chemother. 2015, 70, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, R.P.; Dannemann, B.; Hoetelmans, R.M. Bedaquiline: A review of human pharmacokinetics and drug-drug interactions. J. Antimicrob. Chemother. 2014, 69, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, F.; Lu, J.; Liu, S.; Dorko, K.; Xie, W.; Ma, X. Bedaquiline metabolism: Enzymes and novel metabolites. Drug Metab. Dispos. Biol. Fate Chem. 2014, 42, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Petrella, S.; Cambau, E.; Chauffour, A.; Andries, K.; Jarlier, V.; Sougakoff, W. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob. Agents Chemother. 2006, 50, 2853–2856. [Google Scholar] [CrossRef] [PubMed]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Villellas, C.; Coeck, N.; Thys, K.; Gevers, T.; Vranckx, L.; Lounis, N.; de Jong, B.C.; Koul, A. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS ONE 2014, 9, e102135. [Google Scholar] [CrossRef] [PubMed]
- Somoskovi, A.; Bruderer, V.; Homke, R.; Bloemberg, G.V.; Bottger, E.C. A mutation associated with clofazimine and bedaquiline cross-resistance in MDR-TB following bedaquiline treatment. Eur. Respir. J. 2015, 45, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, G.V.; Keller, P.M.; Stucki, D.; Trauner, A.; Borrell, S.; Latshang, T.; Coscolla, M.; Rothe, T.; Homke, R.; Ritter, C.; et al. Acquired Resistance to Bedaquiline and Delamanid in Therapy for Tuberculosis. N. Engl. J. Med. 2015, 373, 1986–1988. [Google Scholar] [CrossRef] [PubMed]
- Tantry, S.J.; Markad, S.D.; Shinde, V.; Bhat, J.; Balakrishnan, G.; Gupta, A.K.; Ambady, A.; Raichurkar, A.; Kedari, C.; Sharma, S.; et al. Discovery of Imidazo[1,2-a]pyridine Ethers and Squaramides as Selective and Potent Inhibitors of Mycobacterial Adenosine Triphosphate (ATP) Synthesis. J. Med. Chem. 2017, 60, 1379–1399. [Google Scholar] [CrossRef] [PubMed]
- Black, P.A.; Warren, R.M.; Louw, G.E.; van Helden, P.D.; Victor, T.C.; Kana, B.D. Energy metabolism and drug efflux in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [Google Scholar] [CrossRef] [PubMed]
- Malone, L.; Schurr, A.; Lindh, H.; Mc, K.D.; Kiser, J.S.; Williams, J.H. The effect of pyrazinamide (aldinamide) on experimental tuberculosis in mice. Am. Rev. Tuberc. 1952, 65, 511–518. [Google Scholar] [PubMed]
- Yeager, R.L.; Munroe, W.G.; Dessau, F.I. Pyrazinamide (aldinamide) in the treatment of pulmonary tuberculosis. Am. Rev. Tuberc. 1952, 65, 523–546. [Google Scholar] [PubMed]
- Mc, D.W.; Tompsett, R. Activation of pyrazinamide and nicotinamide in acidic environments in vitro. Am. Rev. Tuberc. 1954, 70, 748–754. [Google Scholar]
- Konno, K.; Nagayama, H.; Oka, S. Nicotinamidase in Mycobacteria: A method for distinguishing bovine type tubercle bacilli from other Mycobacteria. Nature 1959, 184 (Suppl. 22), 1743–1744. [Google Scholar] [CrossRef] [PubMed]
- Miotto, P.; Cabibbe, A.M.; Feuerriegel, S.; Casali, N.; Drobniewski, F.; Rodionova, Y.; Bakonyte, D.; Stakenas, P.; Pimkina, E.; Augustynowicz-Kopec, E.; et al. Mycobacterium tuberculosis pyrazinamide resistance determinants: A multicenter study. mBio 2014, 5, e01819-14. [Google Scholar] [CrossRef] [PubMed]
- Sheen, P.; Requena, D.; Gushiken, E.; Gilman, R.H.; Antiparra, R.; Lucero, B.; Lizarraga, P.; Cieza, B.; Roncal, E.; Grandjean, L.; et al. A multiple genome analysis of Mycobacterium tuberculosis reveals specific novel genes and mutations associated with pyrazinamide resistance. BMC Genom. 2017, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Haagsma, A.C.; Pham, H.; Maaskant, J.J.; Mol, S.; Lill, H.; Bald, D. Pyrazinoic acid decreases the proton motive force, respiratory ATP synthesis activity, and cellular ATP levels. Antimicrob. Agents Chemother. 2011, 55, 5354–5357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Njire, M.; Wang, N.; Wang, B.; Tan, Y.; Cai, X.; Liu, Y.; Mugweru, J.; Guo, J.; Hameed, H.M.A.; Tan, S.; et al. Pyrazinoic Acid Inhibits a Bifunctional Enzyme in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00070-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Scorpio, A.; Nikaido, H.; Sun, Z. Role of acid pH and deficient efflux of pyrazinoic acid in unique susceptibility of Mycobacterium tuberculosis to pyrazinamide. J. Bacteriol. 1999, 181, 2044–2049. [Google Scholar] [PubMed]
- Zhang, Y.; Permar, S.; Sun, Z. Conditions that may affect the results of susceptibility testing of Mycobacterium tuberculosis to pyrazinamide. J. Med. Microbiol. 2002, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar] [PubMed]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, N.; Dawson, R.; du Bois, J.; Narunsky, K.; Horwith, G.; Phipps, A.J.; Nacy, C.A.; Aarnoutse, R.E.; Boeree, M.J.; Gillespie, S.H.; et al. Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 2015, 70, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Boeree, M.J.; Heinrich, N.; Aarnoutse, R.; Diacon, A.H.; Dawson, R.; Rehal, S.; Kibiki, G.S.; Churchyard, G.; Sanne, I.; Ntinginya, N.E.; et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: A multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis. 2017, 17, 39–49. [Google Scholar] [CrossRef]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E., 3rd; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.; Kordulakova, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Makobongo, M.O.; Einck, L.; Peek, R.M., Jr.; Merrell, D.S. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. PLoS ONE 2013, 8, e68917. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Santos, P.; Li, K.; Lameira, L.; de Carvalho, T.M.; Huang, G.; Galizzi, M.; Shang, N.; Li, Q.; Gonzalez-Pacanowska, D.; Hernandez-Rodriguez, V.; et al. SQ109, a new drug lead for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of sq109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Properties | Structure |
|---|---|---|---|
| Thioridazine | NDH-2 | MIC90-8–15 µg/mL Approved |  |
| Trifluoperazine | NDH-2 | MIC90-19.2 µg/mL |  |
| Clofazimine | NDH-2 | MIC90-0.25 µg/mL Phase III clinical trials |  |
| Quinolinyl pyrimidine | NDH-2 | MIC90-8-32 µg/mL |  |
| 3-Nitropropionate (3NP) | SDH | MIC90-3.3 µM Active in vivo |  |
| DG70 | MenG | MIC90-4.8 µg/mL |  |
| Aurachin RE | MenA | MIC90 < 12.5 µg/mL |  |
| Ro 48-8071 | MenA | MIC90-3 µg/mL |  |
| 7-Methoxy-2-naphthol | MenA | MIC90-3 µg/mL |  |
| Allylaminomethanone | MenA | MIC90-1 µg/mL |  |
| Aurachin D | Cytochrome bd oxidase | MIC90-85 µM |  |
| PABS (Phenoxyalkylbenzimidazoles) | QcrB | MIC90-0.056 µM |  |
| Q203 (imidazopyridine amide) | QcrB | MIC50-0.28 nM ex vivo MIC50-2.7 nM in vitro Phase I clinical trials |  |
| Pyrrolo[3,4-c]pyridine-1,3-dione | QcrB |  | |
| Lansoprazole | QcrB | MIC90-104 µg/mL Active in vivo |  |
| Bedaquiline | ATP synthase | MIC90-0.004-0.13 µg/mL Approved for MDR-TB Phase III clinical trials for DS-TB |  |
| Squaramide | ATP synthase | MIC90-0.5 µM Active in vivo Pre-clinical trials |  |
| SQ109 | PMF | MIC90-0.78 µg/mL Phase II clinical trials |  |
| Pyrazinamide | PMF | MIC90-100 µg/mL Approved |  |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, I.K.; Bajeli, S.; Akela, A.K.; Kumar, A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens 2018, 7, 24. https://doi.org/10.3390/pathogens7010024
Iqbal IK, Bajeli S, Akela AK, Kumar A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens. 2018; 7(1):24. https://doi.org/10.3390/pathogens7010024
Chicago/Turabian StyleIqbal, Iram Khan, Sapna Bajeli, Ajit Kumar Akela, and Ashwani Kumar. 2018. "Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery" Pathogens 7, no. 1: 24. https://doi.org/10.3390/pathogens7010024
APA StyleIqbal, I. K., Bajeli, S., Akela, A. K., & Kumar, A. (2018). Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens, 7(1), 24. https://doi.org/10.3390/pathogens7010024