The Structure of PrPSc Prions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
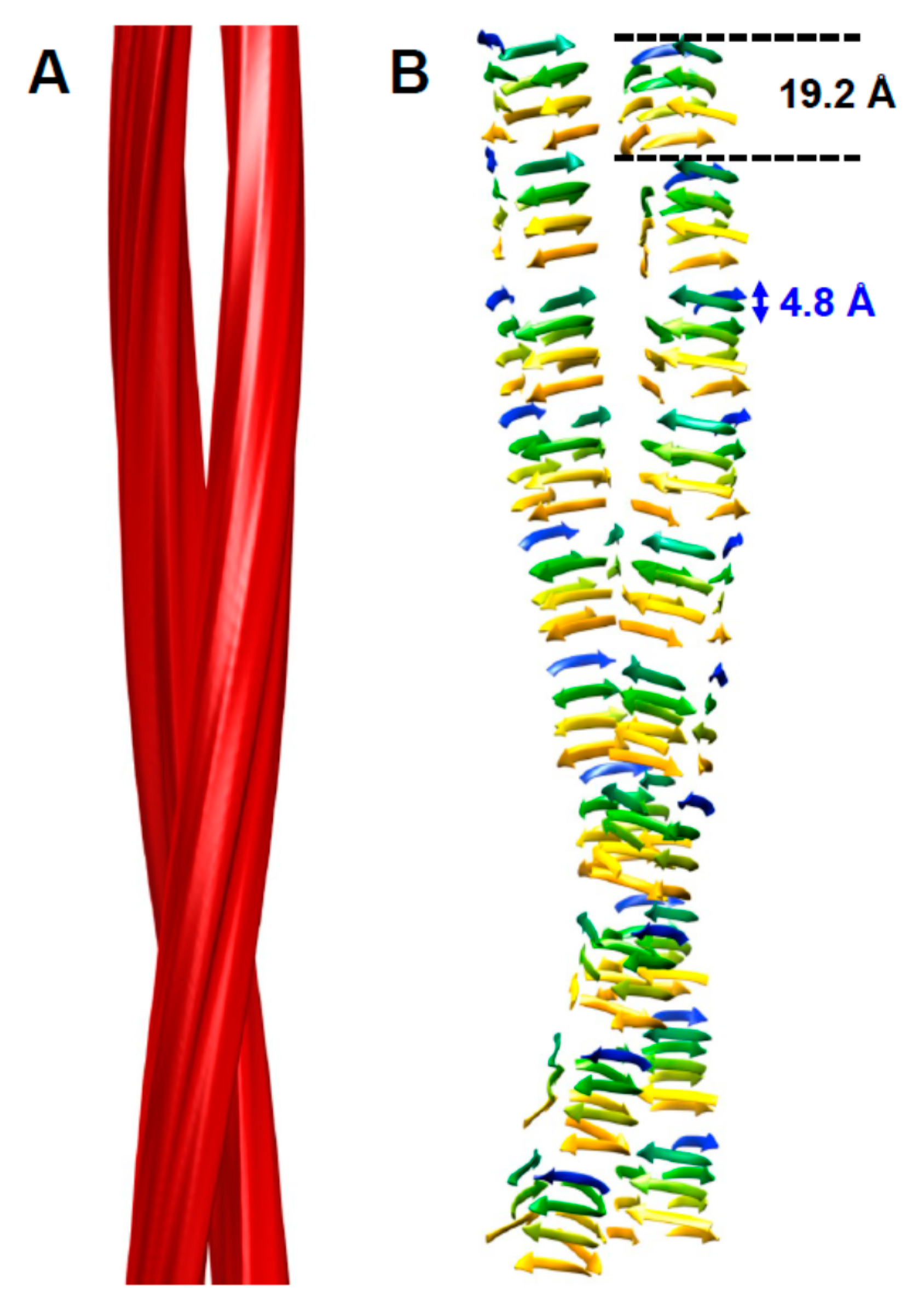
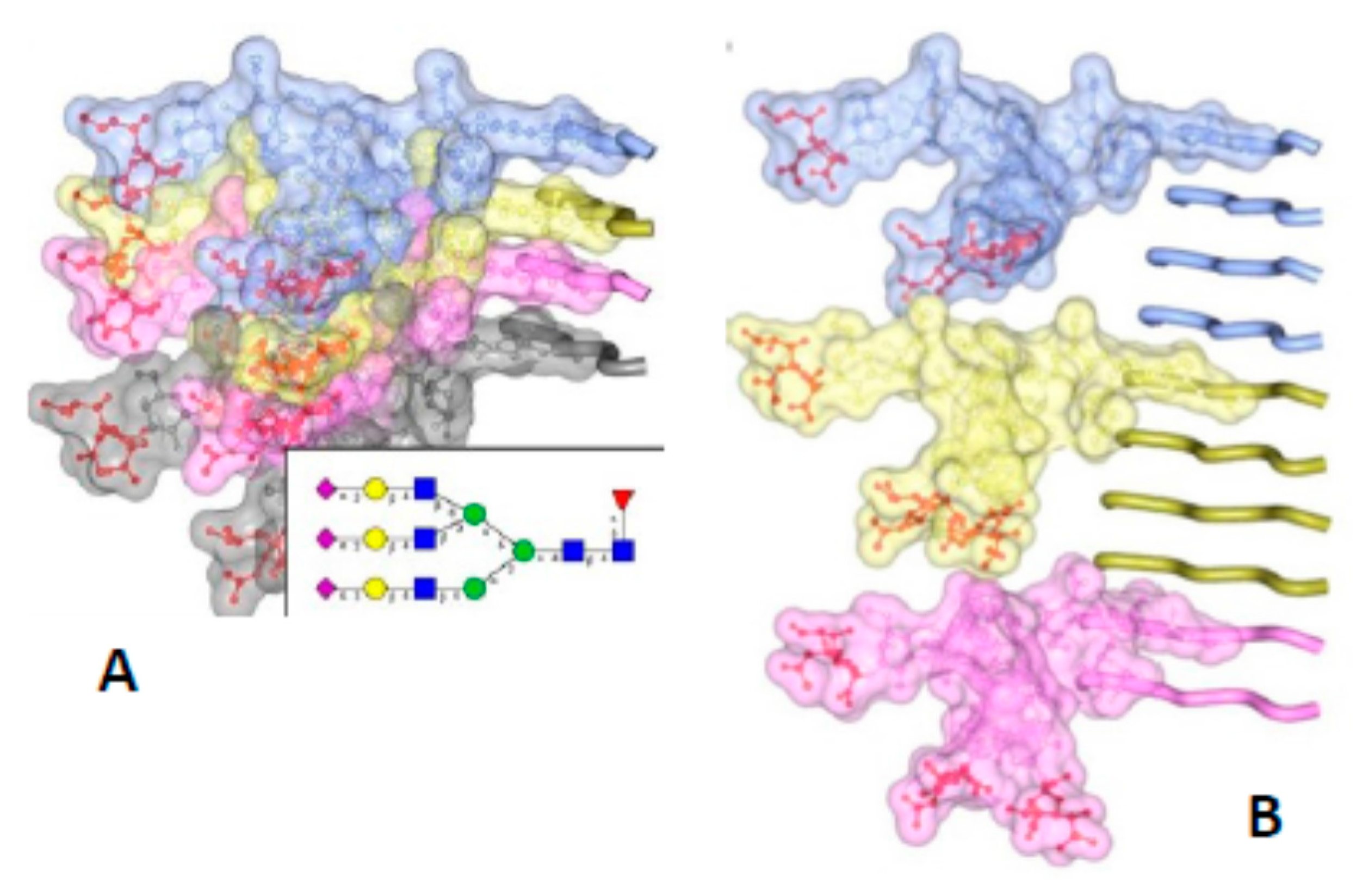
2. The Architecture of PrPSc Prions
3. Other Models of PrPSc
4. Implications of the Structure of PrPSc for Its Propagation
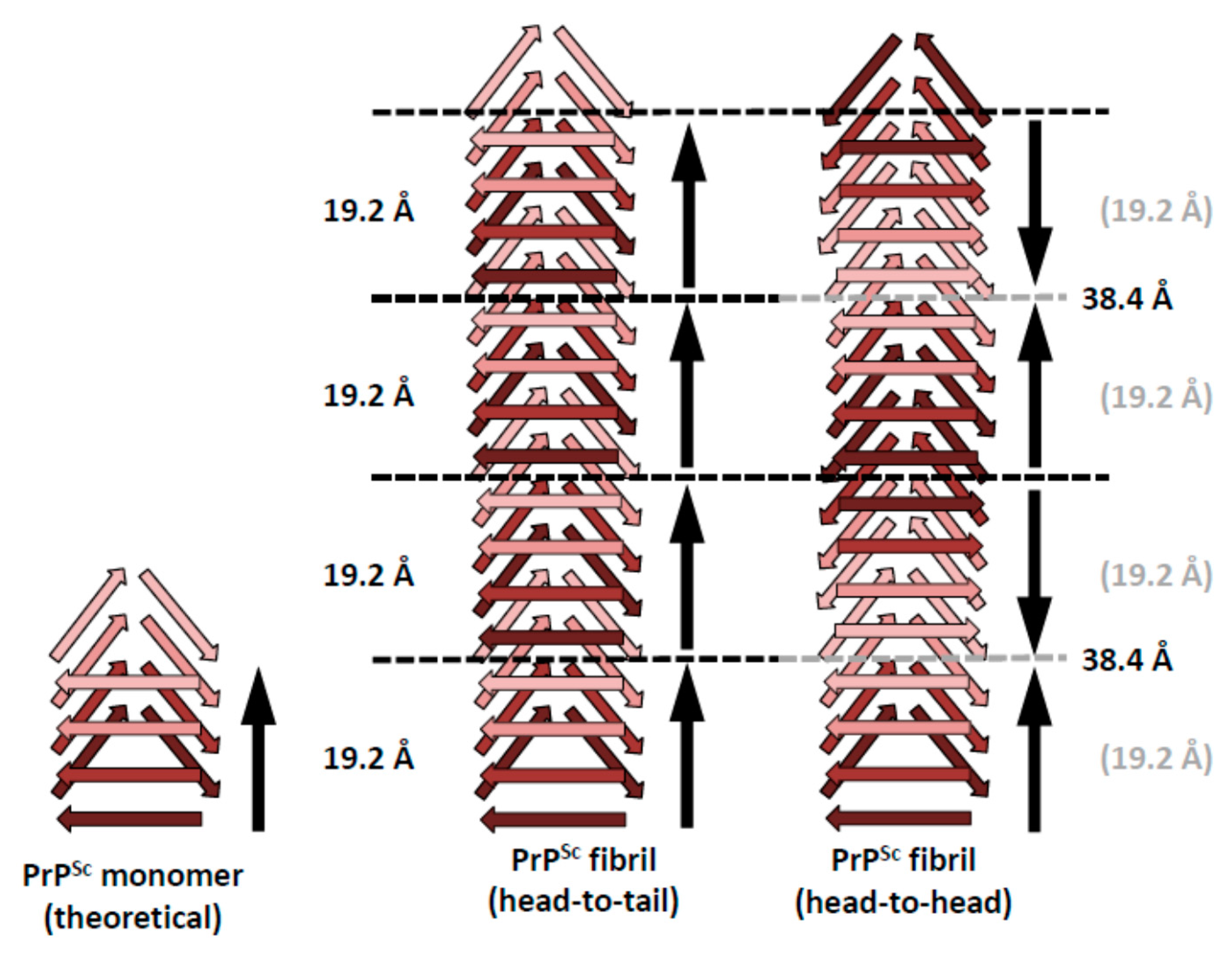
5. Head-to-Head or Head-to-Tail Stacking?
6. Concluding Remarks and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Requena, J.R.; Kristensson, K.; Korth, C.; Zurzolo, C.; Simmons, M.; Aguilar-Calvo, P.; Aguzzi, A.; Andreoletti, O.; Benestad, S.L.; Böhm, R.; et al. The Priority position paper: Protecting Europe’s food chain from prions. Prion 2016, 10, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Cuille, J.; Chelle, P.L. Pathologie animale—La maladie dite tremblante du mouton est-elle inoculable? C. R. Hebd. Seances Acad. Sci. 1936, 203, 1552–1554. [Google Scholar]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef] [PubMed]
- De Marco, M.F.; Linehan, J.; Gill, O.N.; Clewley, J.P.; Brandner, S. Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J. Pathol. 2010, 222, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Alpers, M.P. The epidemiology of kuru: Monitoring the epidemic from its peak to its end. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3707–3713. [Google Scholar] [CrossRef] [PubMed]
- Urwin, P.J.; Mackenzie, J.M.; Llewelyn, C.A.; Will, R.G.; Hewitt, P.E. Creutzfeldt-Jakob disease and blood transfusion: Updated results of the UK Transfusion Medicine Epidemiology Review Study. Vox Sang. 2016, 110, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Hannaoui, S.; Schatzl, H.M.; Gilch, S. Chronic wasting disease: Emerging prions and their potential risk. PLoS Pathog. 2017, 13, e1006619. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikøren, T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet. Res. 2016, 47, 88. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Requena, J.R. Prion-like diseases: Looking for their niche in the realm of infectious diseases. Virus Res. 2015, 207, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Fernández, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The structural architecture of an infectious mammalian prion using electron cryomicroscopy. PLoS Pathog. 2016, 8, e1005835. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Baxa, U.; Steven, A.C. Structural dependence of HET-s amyloid fibril infectivity assessed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 3252–3257. [Google Scholar] [CrossRef] [PubMed]
- Wille, H.; Michelitsch, M.D.; Guenebaut, V.; Supattapone, S.; Serban, A.; Cohen, F.E.; Agard, D.A.; Prusiner, S.B. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. USA 2002, 99, 3563–3568. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, C.; Wille, H.; Prusiner, S.B.; Cohen, F.E. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc. Natl. Acad. Sci. USA 2004, 101, 8342–8347. [Google Scholar] [CrossRef] [PubMed]
- Supattapone, S.; Bosque, P.; Muramoto, T.; Wille, H.; Aagaard, C.; Peretz, D.; Nguyen, H.O.; Heinrich, C.; Torchia, M.; Safar, J.; et al. Prion protein of 106 residues creates an artificial transmission barrier for prion replication in transgenic mice. Cell 1999, 96, 869–878. [Google Scholar] [CrossRef]
- Smirnovas, V.; Baron, G.S.; Offerdahl, D.K.; Raymond, G.J.; Caughey, B.; Surewicz, W.K. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2011, 18, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.R.; Wille, H. The structure of the infectious prion protein: Experimental data and molecular models. Prion 2014, 8, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Wille, H.; Bian, W.; McDonald, M.; Kendall, A.; Colby, D.W.; Bloch, L.; Ollesch, J.; Borovinskiy, A.L.; Cohen, F.E.; Prusiner, S.B.; et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Natl. Acad. Sci. USA 2009, 106, 16990–16995. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Sawaya, M.R.; Makarava, N.; Savtchenko, R.; Nilsson, K.P.; Eisenberg, D.; Baskakov, I.V. Two amyloid States of the prion protein display significantly different folding patterns. J. Mol. Biol. 2010, 400, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Wille, H.; Stöhr, J.; Baxa, U.; Prusiner, S.B.; Stubbs, G. Degradation of fungal prion HET-s (218–289) induces formation of a generic amyloid fold. Biophys. J. 2012, 102, 2339–2344. [Google Scholar] [CrossRef] [PubMed]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s(218–289) prion form a β solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Wille, H.; Stöhr, J.; Kendall, A.; Bian, W.; McDonald, M.; Tiggelaar, S.; Watts, J.C.; Prusiner, S.B.; Stubbs, G. Structural studies of truncated forms of the prion protein PrP. Biophys. J. 2015, 108, 548–1554. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J.; Bessen, R.A. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998, 273, 32230–32235. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Roller, P.P.; Gajdusek, D.C.; Gibbs, C.J., Jr. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J. Biol. Chem. 1993, 268, 20276–20284. [Google Scholar] [PubMed]
- Schmitz, M.; Dittmar, K.; Llorens, F.; Gelpi, E.; Ferrer, I.; Schulz-Schaeffer, W.J.; Zerr, I. Hereditary human prion diseases: An update. Mol. Neurobiol. 2016, 54, 4138–4149. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.Q.; Capellari, S.; Parchi, P.; Sy, M.S.; Gambetti, P.; Chen, S.G. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J. Biol. Chem. 2003, 278, 40429–40436. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Farinazzo, A.; Prelli, F.; Fiorini, M.; Gelati, M.; Ferrari, S.; Righetti, P.G.; Rizzuto, N.; Frangione, B.; Monaco, S. Identification of distinct N-terminal truncated forms of prion protein in different Creutzfeldt-Jakob disease subtypes. J. Biol. Chem. 2004, 279, 38936–38942. [Google Scholar] [CrossRef] [PubMed]
- Sajnani, G.; Pastrana, M.A.; Dynin, I.; Onisko, B.; Requena, J.R. Scrapie prion protein structural constraints obtained by limited proteolysis and mass spectrometry. J. Mol. Biol. 2008, 382, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Fernández, E.; Alonso, J.; Pastrana, M.A.; Ramos, A.; Stitz, L.; Vidal, E.; Dynin, I.; Petsch, B.; Silva, C.J.; Requena, J.R. Structural organization of mammalian prions as probed by limited proteolysis. PLoS ONE 2012, 7, e50111. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.J. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1998, 1382, 191–206. [Google Scholar] [CrossRef]
- Silva, C.J.; Vázquez-Fernández, E.; Onisko, B.; Requena, J.R. Proteinase K and the structure of PrPSc: The good, the bad and the ugly. Virus Res. 2015, 207, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Gambetti, P.; Dong, Z.; Yuan, J.; Xiao, X.; Zheng, M.; Alshekhlee, A.; Castellani, R.; Cohen, M.; Barria, M.A.; Gonzalez-Romero, D.; et al. A novel human disease with abnormal prion protein sensitive to protease. Ann. Neurol. 2008, 63, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Pirisinu, L.; Nonno, R.; Esposito, E.; Benestad, S.L.; Gambetti, P.; Agrimi, U.; Zou, W.Q. Small ruminant Nor98 prions share biochemical features with human Gerstmann-Straussler-Scheinker disease and variably protease-sensitive prionopathy. PLoS ONE 2013, 8, e66405. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Stöhr, J.; Oehler, A.; Bhardwaj, S.; Grillo, S.K.; Patel, S.; DeArmond, S.J.; Prusiner, S.B. Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc. Natl. Acad. Sci. USA 2012, 109, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Bourkas, M.E.; Patel, S.; Oehler, A.; Gavidia, M.; Bhardwaj, S.; Lee, J.; Prusiner, S.B. Towards authentic transgenic mouse models of heritable PrP prion diseases. Acta Neuropathol. 2016, 132, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Götte, D.R.; Benestad, S.L.; Laude, H.; Zurbriggen, A.; Oevermann, A.; Seuberlich, T. Atypical scrapie isolates involve a uniform prion species with a complex molecular signature. PLoS ONE 2011, 6, e27510. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Dolan, M.A.; Taubner, L.M.; Kraus, A.; Wickner, R.B.; Caughey, B. Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J. Biol. Chem. 2014, 289, 24129–24142. [Google Scholar] [CrossRef] [PubMed]
- Sim, V.L.; Caughey, B. Ultrastructures and strain comparison of under-glycosylated scrapie prion fibrils. Neurobiol. Aging 2009, 30, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V.; Katorcha, E. Multifaceted role of sialylation in prion diseases. Front. Neurosci. 2016, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Fernández, E.; Young, H.S.; Requena, J.R.; Wille, H. The structure of mammalian prions and their aggregates. Int. Rev. Cell Mol. Biol. 2017, 329, 277–301. [Google Scholar] [PubMed]
- Richardson, J.S.; Richardson, D.C. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.W.; Starner-Kreinbrink, J.L.; Hosur, R.; Clark, P.L.; Berger, B. Structure-based prediction reveals capping motifs that inhibit β-helix aggregation. Proc. Natl. Acad. Sci. USA 2011, 108, 11099–11104. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, J.P.; Fuentes, G.; Boshuizen, R.; Bonvin, A.M. Two-rung model of a left-handed beta-helix for prions explains species barrier and strain variation in transmissible spongiform encephalopathies. J. Mol. Biol. 2006, 360, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Stura, E.A.; Muller, B.H.; Bossus, M.; Michel, S.; Jolivet-Reynaud, C.; Ducancel, F. Crystal structure of human prostate-specific antigen in a sandwich antibody complex. J. Mol. Biol. 2011, 414, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Friedmann, M.P.; Riek, R. Amyloid aggregates arise from amino acid condensations under prebiotic conditions. Angew. Chem. Int. Ed. 2016, 55, 11609–11613. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wille, H.; Requena, J.R. The Structure of PrPSc Prions. Pathogens 2018, 7, 20. https://doi.org/10.3390/pathogens7010020
Wille H, Requena JR. The Structure of PrPSc Prions. Pathogens. 2018; 7(1):20. https://doi.org/10.3390/pathogens7010020
Chicago/Turabian StyleWille, Holger, and Jesús R. Requena. 2018. "The Structure of PrPSc Prions" Pathogens 7, no. 1: 20. https://doi.org/10.3390/pathogens7010020
APA StyleWille, H., & Requena, J. R. (2018). The Structure of PrPSc Prions. Pathogens, 7(1), 20. https://doi.org/10.3390/pathogens7010020