
Immunomodulation and Disease Tolerance to Staphylococcus aureus

{kind=link}
Abstract
:1. Introduction
2. Innate and Adaptive Immune Responses to S. aureus
2.1. Innate Immunity to S. aureus
2.2. Adaptive Immunity to S. aureus
3. Cellular Basis of Immunomodulation by S. aureus
4. Molecular Mechanisms of Immunomodulation by S. aureus
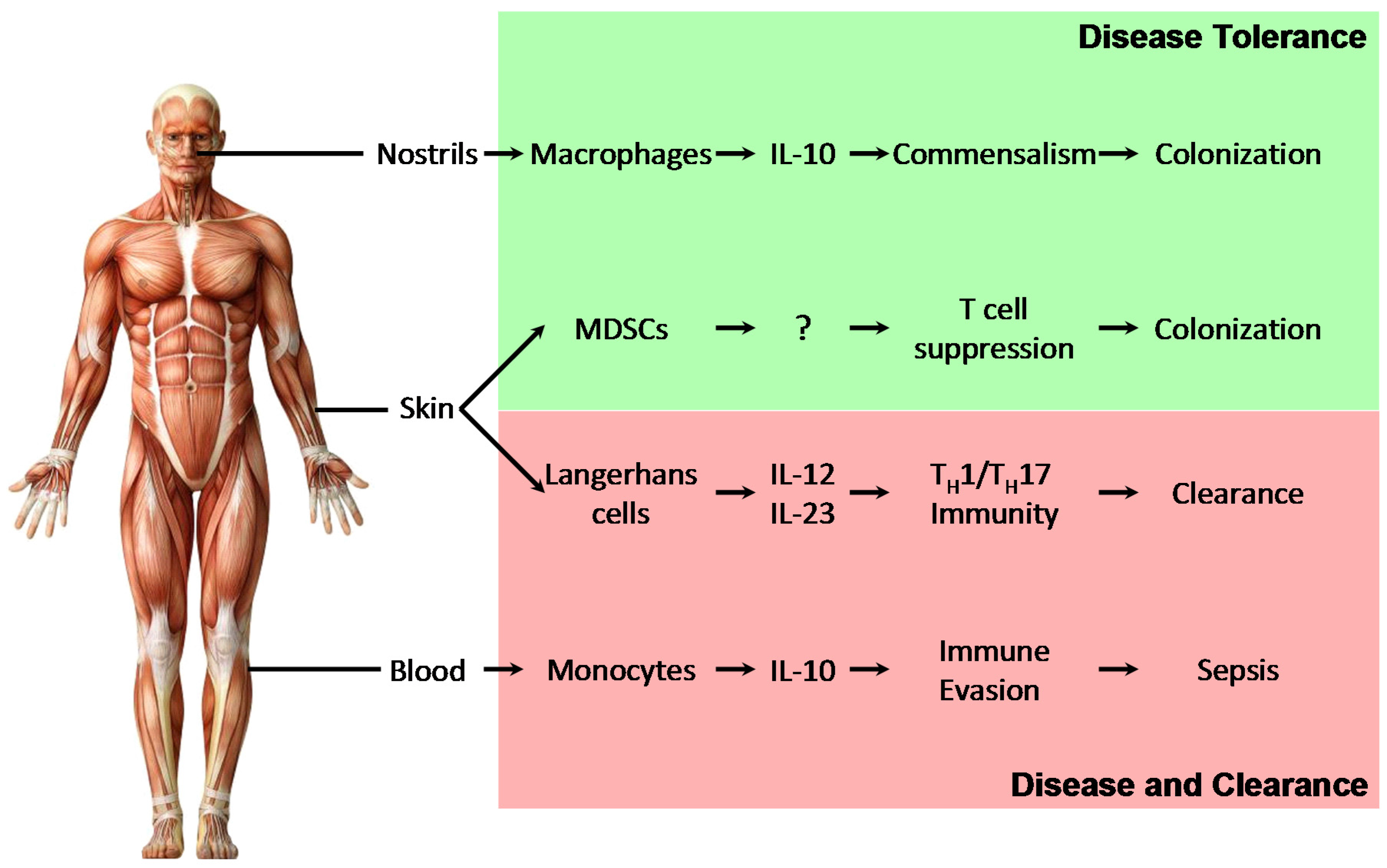
5. Mechanisms of Disease Tolerance to S. aureus
6. Mechanisms of Immune Evasion by S. aureus
7. Clinical Implications and Future Prospects
8. Conclusions
Acknowledgments
Author Contribution
Conflicts of Interest
References
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.F.; Leech, J.M.; Rogers, T.R.; McLoughlin, R.M. Staphylococcus aureus colonization: Modulation of host immune response and impact on human vaccine design. Front. Immunol. 2014, 4, 507. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F. The changing epidemiology of methicillin-resistant Staphylococcus aureus: 50 years of a superbug. Am. J. Infect. Control 2013, 41, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Cho, J.S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 2011, 11, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Dantes, R.; Mu, Y.; Belflower, R.; Aragon, D.; Dumyati, G.; Harrison, L.H.; Lessa, F.C.; Lynfield, R.; Nadle, J.; Petit, S.; et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, united states, 2011. JAMA Intern. Med. 2013, 173, 1970–1978. [Google Scholar] [PubMed]
- Stentzel, S.; Sundaramoorthy, N.; Michalik, S.; Nordengrun, M.; Schulz, S.; Kolata, J.; Kloppot, P.; Engelmann, S.; Steil, L.; Hecker, M.; et al. Specific serum igg at diagnosis of Staphylococcus aureus bloodstream invasion is correlated with disease progression. J. Proteom. 2015, 128, 1–7. [Google Scholar]
- Wertheim, H.F.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Bar-Gal, G.K.; Blum, S.E.; Hadas, L.; Ehricht, R.; Monecke, S.; Leitner, G. Host-specificity of Staphylococcus aureus causing intramammary infections in dairy animals assessed by genotyping and virulence genes. Vet. Microbiol. 2015, 176, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Pishchany, G.; McCoy, A.L.; Torres, V.J.; Krause, J.C.; Crowe, J.E., Jr.; Fabry, M.E.; Skaar, E.P. Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe 2010, 8, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Peres, A.G.; Madrenas, J. The broad landscape of immune interactions with Staphylococcus aureus: From commensalism to lethal infections. Burns 2013, 39, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, H.F.; Vos, M.C.; Ott, A.; van Belkum, A.; Voss, A.; Kluytmans, J.A.; van Keulen, P.H.; Vandenbroucke-Grauls, C.M.; Meester, M.H.; Verbrugh, H.A. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet 2004, 364, 703–705. [Google Scholar] [CrossRef]
- Netsvyetayeva, I.; Fraczek, M.; Piskorska, K.; Golas, M.; Sikora, M.; Swoboda-Kopec, E.; Mlynarczyk, A.; Marusza, W.; Palmieri, B.; Iannitti, T. Staphylococcus aureus nasal carriage in ukraine: Antibacterial resistance and virulence factor encoding genes. BMC Infect. Dis. 2014, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and langerhans cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Frodermann, V.; Chau, T.A.; Sayedyahossein, S.; Toth, J.M.; Heinrichs, D.E.; Madrenas, J. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis 2011, 204, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Skabytska, Y.; Wolbing, F.; Gunther, C.; Koberle, M.; Kaesler, S.; Chen, K.M.; Guenova, E.; Demircioglu, D.; Kempf, W.E.; Volz, T.; et al. Cutaneous innate immune sensing of toll-like receptor 2–6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity 2014, 41, 762–775. [Google Scholar] [PubMed]
- Takeuchi, O.; Hoshino, K.; Akira, S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 2000, 165, 5392–5396. [Google Scholar] [CrossRef] [PubMed]
- Von Bernuth, H.; Picard, C.; Jin, Z.; Pankla, R.; Xiao, H.; Ku, C.L.; Chrabieh, M.; Mustapha, I.B.; Ghandil, P.; Camcioglu, Y.; et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 2008, 321, 691–696. [Google Scholar] [PubMed]
- Alsina, L.; Israelsson, E.; Altman, M.C.; Dang, K.K.; Ghandil, P.; Israel, L.; von Bernuth, H.; Baldwin, N.; Qin, H.; Jin, Z.; et al. A narrow repertoire of transcriptional modules responsive to pyogenic bacteria is impaired in patients carrying loss-of-function mutations in MyD88 or IRAK4. Nat. Immunol. 2014, 15, 1134–1142. [Google Scholar] [PubMed]
- Tomlinson, G.; Chimalapati, S.; Pollard, T.; Lapp, T.; Cohen, J.; Camberlein, E.; Stafford, S.; Periselneris, J.; Aldridge, C.; Vollmer, W.; et al. TLR-mediated inflammatory responses to streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J. Immunol. 2014, 193, 3736–3745. [Google Scholar] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Peres, A.G.; Stegen, C.; Li, J.; Xu, A.Q.; Levast, B.; Surette, M.G.; Cousineau, B.; Desrosiers, M.; Madrenas, J. Uncoupling of pro- and anti-inflammatory properties of Staphylococcus aureus. Infect. Immun. 2015, 83, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Greenlee-Wacker, M.C.; Rigby, K.M.; Kobayashi, S.D.; Porter, A.R.; DeLeo, F.R.; Nauseef, W.M. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J. Immunol. 2014, 192, 4709–4717. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, R.M.; Lee, J.C.; Kasper, D.L.; Tzianabos, A.O. Ifn-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. J. Immunol. 2008, 181, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Daniels, M.; Zhao, F.; Alegre, M.L.; Chong, A.S.; Daum, R.S. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17a. Infect. Immun. 2014, 82, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(h)17 cell differentiation in subjects with autosomal dominant hyper-ige syndrome. Nature 2008, 452, 773–776. [Google Scholar]
- Soltesz, B.; Toth, B.; Sarkadi, A.K.; Erdos, M.; Marodi, L. The evolving view of IL-17-mediated immunity in defense against mucocutaneous candidiasis in humans. Int. Rev. Immunol. 2015, 34, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Al-Muhsen, S.; Halwani, R.; Ma, C.S.; et al. Immunodeficiencies. Impairment of immunity to candida and mycobacterium in humans with bi-allelic rorc mutations. Science 2015, 349, 606–613. [Google Scholar] [PubMed]
- Grumann, D.; Scharf, S.S.; Holtfreter, S.; Kohler, C.; Steil, L.; Engelmann, S.; Hecker, M.; Volker, U.; Broker, B.M. Immune cell activation by enterotoxin gene cluster (egc)-encoded and non-egc superantigens from Staphylococcus aureus. J. Immunol. 2008, 181, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Dauwalder, O.; Thomas, D.; Ferry, T.; Debard, A.L.; Badiou, C.; Vandenesch, F.; Etienne, J.; Lina, G.; Monneret, G. Comparative inflammatory properties of staphylococcal superantigenic enterotoxins sea and seg: Implications for septic shock. J. Leukoc Biol. 2006, 80, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Kolata, J.B.; Kuhbandner, I.; Link, C.; Normann, N.; Vu, C.H.; Steil, L.; Weidenmaier, C.; Broker, B.M. The fall of a dogma? Unexpected high T-cell memory response to Staphylococcus aureus in humans. J. Infect. Dis. 2015, 212, 830–838. [Google Scholar] [PubMed]
- Schlievert, P.M. The dream of staphylococcal vaccination. J. Exp. Med. 2014, 211, 2326. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Avalos, A.M.; Ploegh, H.L. Accessory molecules for toll-like receptors and their function. Nat. Rev. Immunol. 2012, 12, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.; Chaffois, C.; Gaillard, C.; Saulnier, V.; Merck, E.; Tancredi, S.; Guiet, C.; Briere, F.; Vlach, J.; Lebecque, S.; et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J. Immunol. 2005, 174, 2942–2950. [Google Scholar]
- Oosting, M.; Cheng, S.C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; van Eenennaam, H.; Sturm, P.; et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Friedrich, A.W.; Lubritz, G.; Weilert, M.; Peters, G.; von Eiff, C. Prevalence of genes encoding pyrogenic toxin superantigens and exfoliative toxins among strains of Staphylococcus aureus isolated from blood and nasal specimens. J. Clin. Microbiol. 2003, 41, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Mempel, M.; Lina, G.; Hojka, M.; Schnopp, C.; Seidl, H.P.; Schafer, T.; Ring, J.; Vandenesch, F.; Abeck, D. High prevalence of superantigens associated with the egc locus in Staphylococcus aureus isolates from patients with atopic eczema. Eur. J. Clin. Microbiol. Infect. Dis. 2003, 22, 306–309. [Google Scholar] [PubMed]
- Nashev, D.; Toshkova, K.; Salasia, S.I.; Hassan, A.A.; Lammler, C.; Zschock, M. Distribution of virulence genes of Staphylococcus aureus isolated from stable nasal carriers. FEMS Microbiol. Lett. 2004, 233, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.P.; Chesney, P.J.; Wand, P.J.; LaVenture, M. Toxic-shock syndrome: Epidemiologic features, recurrence, risk factors, and prevention. N. Engl. J. Med. 1980, 303, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Reduced incidence of menstrual toxic-shock syndrome--United States, 1980–1990. MMWR Morb. Mortal Wkly Rep. 1990, 39, 424–423.
- Chau, T.A.; McCully, M.L.; Brintnell, W.; An, G.; Kasper, K.J.; Vines, E.D.; Kubes, P.; Haeryfar, S.M.; McCormick, J.K.; Cairns, E.; et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat. Med. 2009, 15, 641–648. [Google Scholar] [PubMed]
- Lai, Y.; di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.R.; Hooper, L.V.; Schmidt, R.R.; von Aulock, S.; et al. Commensal bacteria regulate toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [PubMed]
- Tkaczyk, C.; Hamilton, M.M.; Datta, V.; Yang, X.P.; Hilliard, J.J.; Stephens, G.L.; Sadowska, A.; Hua, L.; O’Day, T.; Suzich, J.; et al. Staphylococcus aureus alpha toxin suppresses effective innate and adaptive immune responses in a murine dermonecrosis model. PLoS ONE 2013, 8, e75103. [Google Scholar]
- Hawrylowicz, C.M.; O’Garra, A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat. Rev. Immunol. 2005, 5, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Sendide, K.; Deghmane, A.E.; Pechkovsky, D.; Av-Gay, Y.; Talal, A.; Hmama, Z. Mycobacterium bovis bcg attenuates surface expression of mature class II molecules through IL-10-dependent inhibition of cathepsin s. J. Immunol. 2005, 175, 5324–5332. [Google Scholar] [CrossRef] [PubMed]
- Willems, F.; Marchant, A.; Delville, J.P.; Gerard, C.; Delvaux, A.; Velu, T.; de Boer, M.; Goldman, M. Interleukin-10 inhibits B7 and intercellular adhesion molecule-1 expression on human monocytes. Eur. J. Immunol. 1994, 24, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Buelens, C.; Willems, F.; Delvaux, A.; Pierard, G.; Delville, J.P.; Velu, T.; Goldman, M. Interleukin-10 differentially regulates B7–1 (CD80) and B7–2 (CD86) expression on human peripheral blood dendritic cells. Eur. J. Immunol. 1995, 25, 2668–2672. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Heim, C.E.; Angle, A.; Sanderson, S.D.; Kielian, T. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J. Immunol. 2013, 190, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Holtappels, G.; Zhang, N.; Kubica, M.; Deswarte, K.; Derycke, L.; Claeys, S.; Hammad, H.; Brusselle, G.G.; Vandenabeele, P.; et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy 2011, 66, 396–403. [Google Scholar] [PubMed]
- Xu, F.; Kang, Y.; Zhang, H.; Piao, Z.; Yin, H.; Diao, R.; Xia, J.; Shi, L. Akt1-mediated regulation of macrophage polarization in a murine model of Staphylococcus aureus pulmonary infection. J. Infect Dis. 2013, 208, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D. Expression and function of toll-like receptors in T lymphocytes. Curr. Opin. Immunol. 2007, 19, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human Th17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Rabe, H.; Nordstrom, I.; Andersson, K.; Lundell, A.C.; Rudin, A. Staphylococcus aureus convert neonatal conventional cd4(+) T cells into Foxp3(+) CD25(+) CD127(low) T cells via the PD-1/PD-lL axis. Immunology 2014, 141, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.E.; Eickhoff, J.C.; Shukla, S.K.; Pantrangi, M.; Rooijakkers, S.; Cosgrove, S.E.; Nizet, V.; Sakoulas, G. Elevated serum interleukin-10 at time of hospital admission is predictive of mortality in patients with Staphylococcus aureus bacteremia. J. Infect. Dis. 2012, 206, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Tebartz, C.; Horst, S.A.; Sparwasser, T.; Huehn, J.; Beineke, A.; Peters, G.; Medina, E. A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J. Immunol. 2015, 194, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Roderiquez, G.; Norcross, M.A. Control of adaptive immune responses by Staphylococcus aureus through IL-10, PD-L1, and TLR2. Sci. Rep. 2012, 2, 606. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Miyamoto, Y.J.; McIntyre, B.W.; Hook, M.; McCrea, K.W.; McDevitt, D.; Brown, E.L. The Staphylococcus aureus map protein is an immunomodulator that interferes with T cell-mediated responses. J. Clin. Invest. 2002, 110, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Santos-Sierra, S.; Deshmukh, S.D.; Kalnitski, J.; Kuenzi, P.; Wymann, M.P.; Golenbock, D.T.; Henneke, P. Mal connects TLR2 to PI3kinase activation and phagocyte polarization. EMBO J. 2009, 28, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, X.; Edwards, J.P.; Mosser, D.M. Nf-kappab1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 2006, 281, 26041–26050. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Prosch, S.; Schenke-Layland, K.; Riese, U.; Gausmann, U.; Platzer, C. Camp-induced interleukin-10 promoter activation depends on ccaat/enhancer-binding protein expression and monocytic differentiation. J. Biol. Chem. 2003, 278, 5597–5604. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Tseng, H.P.; Chen, L.C.; Chen, B.K.; Chang, W.C. Functional cooperation of simian virus 40 promoter factor 1 and ccaat/enhancer-binding protein beta and delta in lipopolysaccharide-induced gene activation of IL-10 in mouse macrophages. J. Immunol. 2003, 171, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Ananieva, O.; Darragh, J.; Johansen, C.; Carr, J.M.; McIlrath, J.; Park, J.M.; Wingate, A.; Monk, C.E.; Toth, R.; Santos, S.G.; et al. The kinases msk1 and msk2 act as negative regulators of toll-like receptor signaling. Nat. Immunol. 2008, 9, 1028–1036. [Google Scholar] [PubMed]
- Ziegler-Heitbrock, L.; Lotzerich, M.; Schaefer, A.; Werner, T.; Frankenberger, M.; Benkhart, E. Ifn-alpha induces the human IL-10 gene by recruiting both IFN regulatory factor 1 and stat3. J. Immunol. 2003, 171, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Brightbill, H.D.; Plevy, S.E.; Modlin, R.L.; Smale, S.T. A prominent role for sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J. Immunol. 2000, 164, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Tone, M.; Powell, M.J.; Tone, Y.; Thompson, S.A.; Waldmann, H. IL-10 gene expression is controlled by the transcription factors sp1 and sp3. J. Immunol. 2000, 165, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Ruimy, R.; Angebault, C.; Djossou, F.; Dupont, C.; Epelboin, L.; Jarraud, S.; Lefevre, L.A.; Bes, M.; Lixandru, B.E.; Bertine, M.; et al. Are host genetics the predominant determinant of persistent nasal Staphylococcus aureus carriage in humans? J. Infect. Dis. 2010, 202, 924–934. [Google Scholar] [PubMed]
- Alva-Murillo, N.; Tellez-Perez, A.D.; Medina-Estrada, I.; Alvarez-Aguilar, C.; Ochoa-Zarzosa, A.; Lopez-Meza, J.E. Modulation of the inflammatory response of bovine mammary epithelial cells by cholecalciferol (vitamin D) during Staphylococcus aureus internalization. Microb. Pathog. 2014, 77, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.G. TGF-beta in infections and infectious diseases. Microbes Infect. 1999, 1, 1313–1325. [Google Scholar] [CrossRef]
- Neill, D.R.; Coward, W.R.; Gritzfeld, J.F.; Richards, L.; Garcia-Garcia, F.J.; Dotor, J.; Gordon, S.B.; Kadioglu, A. Density and duration of pneumococcal carriage is maintained by transforming growth factor beta1 and T regulatory cells. Am. J. Respir. Crit. Care Med. 2014, 189, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, C.S.; Pereyra, E.A.; Baravalle, C.; Renna, M.S.; Ortega, H.H.; Calvinho, L.F.; Dallard, B.E. Staphylococcus aureus chronic intramammary infection modifies protein expression of transforming growth factor beta (TGF-beta) subfamily components during active involution. Res. Vet. Sci. 2014, 96, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Gozzelino, R.; Weis, S. Tissue damage control in disease tolerance. Trends Immunol. 2014, 35, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Raberg, L.; Sim, D.; Read, A.F. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science 2007, 318, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassu, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [PubMed]
- Figueiredo, N.; Chora, A.; Raquel, H.; Pejanovic, N.; Pereira, P.; Hartleben, B.; Neves-Costa, A.; Moita, C.; Pedroso, D.; Pinto, A.; et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity 2013, 39, 874–884. [Google Scholar] [PubMed]
- Maurer, K.; Reyes-Robles, T.; Alonzo, F., 3rd; Durbin, J.; Torres, V.J.; Cadwell, K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe 2015, 17, 429–440. [Google Scholar]
- Chen, J.Y.; Hour, T.C.; Yang, S.F.; Chien, C.Y.; Chen, H.R.; Tsai, K.L.; Ko, J.Y.; Wang, L.F. Autophagy is deficient in nasal polyps: Implications for the pathogenesis of the disease. Int. Forum. Allergy Rhinol. 2015, 5, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Hermann, I.; Rath, S.; Ziesemer, S.; Volksdorf, T.; Dress, R.J.; Gutjahr, M.; Muller, C.; Beule, A.G.; Hildebrandt, J.P. Staphylococcus aureus hemolysin a disrupts cell-matrix adhesions in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.; Roscioli, E.; Murphy, J.; Ou, J.; Bassiouni, A.; Wormald, P.J.; Vreugde, S. Staphylococcus aureus impairs the airway epithelial barrier in vitro. Int. Forum. Allergy Rhinol. 2015, 5, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Burian, M.; Wolz, C.; Goerke, C. Regulatory adaptation of Staphylococcus aureus during nasal colonization of humans. PLoS ONE 2010, 5, e10040. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.; Diep, B.A.; Mai, T.T.; Vo, N.H.; Warrener, P.; Suzich, J.; Stover, C.K.; Sellman, B.R. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. mBio 2015, 6, e02272–02214. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.; Fishaut, M.; Kapral, F.; Welch, T. Toxic-shock syndrome associated with phage-group-i staphylococci. Lancet 1978, 2, 1116–1118. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Llewelyn, M.; Cohen, J. Superantigens: Microbial agents that corrupt immunity. Lancet Infect. Dis. 2002, 2, 156–162. [Google Scholar] [CrossRef]
- Murray, R.J. Recognition and management of Staphylococcus aureus toxin-mediated disease. Intern. Med. J. 2005, 35 (Suppl. 2), S106–S119. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Clair, B.; Salomon, J. Managing toxic shock syndrome with antibiotics. Expert Opin. Pharmacother. 2004, 5, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [PubMed]
- Lu, J.; Philpott, D.J.; Saunders, P.R.; Perdue, M.H.; Yang, P.C.; McKay, D.M. Epithelial ion transport and barrier abnormalities evoked by superantigen-activated immune cells are inhibited by interleukin-10 but not interleukin-4. J. Pharmacol. Exp. Ther. 1998, 287, 128–136. [Google Scholar] [PubMed]
- Kasper, K.J.; Zeppa, J.J.; Wakabayashi, A.T.; Xu, S.X.; Mazzuca, D.M.; Welch, I.; Baroja, M.L.; Kotb, M.; Cairns, E.; Cleary, P.P.; et al. Bacterial superantigens promote acute nasopharyngeal infection by Streptococcus pyogenes in a human MHC class II-dependent manner. PLoS Pathog. 2014, 10, e1004155. [Google Scholar]
- Muller, B.; de Groot, E.J.; Kortekaas, I.J.; Fokkens, W.J.; van Drunen, C.M. Nasal epithelial cells express IL-10 at levels that negatively correlate with clinical symptoms in patients with house dust mite allergy. Allergy 2007, 62, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; de Groot, E.J.; Kortekaas, I.J.; Fokkens, W.J.; van Drunen, C.M. Nasal endothelial interleukin-10 expression is negatively correlated with nasal symptoms after allergen provocation. Allergy 2009, 64, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [PubMed]
- Okumura, C.Y.; Nizet, V. Subterfuge and sabotage: Evasion of host innate defenses by invasive gram-positive bacterial pathogens. Annu. Rev. Microbiol. 2014, 68, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; Ruyken, M.; Roos, A.; Daha, M.R.; Presanis, J.S.; Sim, R.B.; van Wamel, W.J.; van Kessel, K.P.; van Strijp, J.A. Immune evasion by a staphylococcal complement inhibitor that acts on c3 convertases. Nat. Immunol. 2005, 6, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.S.; Echague, C.G.; Sholl, A.M.; Watkins, J.A.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Clumping factor a interaction with complement factor i increases c3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 2010, 78, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Jongerius, I.; Kohl, J.; Pandey, M.K.; Ruyken, M.; van Kessel, K.P.; van Strijp, J.A.; Rooijakkers, S.H. Staphylococcal complement evasion by various convertase-blocking molecules. J. Exp. Med. 2007, 204, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Jusko, M.; Potempa, J.; Kantyka, T.; Bielecka, E.; Miller, H.K.; Kalinska, M.; Dubin, G.; Garred, P.; Shaw, L.N.; Blom, A.M. Staphylococcal proteases aid in evasion of the human complement system. J. Innate Immun. 2014, 6, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.J.; Ruyken, M.; Malone, C.L.; van Strijp, J.A.; Horswill, A.R.; Rooijakkers, S.H. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J. Immunol. 2011, 186, 6445–6453. [Google Scholar] [CrossRef] [PubMed]
- De Haas, C.J.; Veldkamp, K.E.; Peschel, A.; Weerkamp, F.; van Wamel, W.J.; Heezius, E.C.; Poppelier, M.J.; van Kessel, K.P.; van Strijp, J.A. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J. Exp. Med. 2004, 199, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Thakker, M.; Park, J.S.; Carey, V.; Lee, J.C. Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect. Immun. 1998, 66, 5183–5189. [Google Scholar] [PubMed]
- Watts, A.; Ke, D.; Wang, Q.; Pillay, A.; Nicholson-Weller, A.; Lee, J.C. Staphylococcus aureus strains that express serotype 5 or serotype 8 capsular polysaccharides differ in virulence. Infect. Immun. 2005, 73, 3502–3511. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.; Sokolovska, A.; Charriere, G.M.; Boyer, L.; Dejardin, S.; Cappillino, M.P.; Yantosca, L.M.; Takahashi, K.; Moore, K.J.; Lacy-Hulbert, A.; et al. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J. Immunol. 2010, 184, 7071–7081. [Google Scholar] [PubMed]
- Shi, L.; Takahashi, K.; Dundee, J.; Shahroor-Karni, S.; Thiel, S.; Jensenius, J.C.; Gad, F.; Hamblin, M.R.; Sastry, K.N.; Ezekowitz, R.A. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J. Exp. Med. 2004, 199, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Falugi, F.; Kim, H.K.; Missiakas, D.M.; Schneewind, O. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Mandell, G.L. Catalase, superoxide dismutase, and virulence of Staphylococcus aureus. In vitro and in vivo studies with emphasis on staphylococcal--leukocyte interaction. J. Clin. Invest. 1975, 55, 561–566. [Google Scholar] [PubMed]
- Karavolos, M.H.; Horsburgh, M.J.; Ingham, E.; Foster, S.J. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology 2003, 149, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Biswas, R.; Herbert, S.; Kulauzovic, E.; Weidenmaier, C.; Peschel, A.; Gotz, F. Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J. Bacteriol. 2007, 189, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Park, B.G.; Wolf, A.J.; Brikos, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Gotz, F.; et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 2010, 7, 38–49. [Google Scholar] [PubMed]
- O’Leary, S.; O’Sullivan, M.P.; Keane, J. IL-10 blocks phagosome maturation in mycobacterium tuberculosis-infected human macrophages. Am. J. Respir. Cell Mol. Biol. 2011, 45, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, K.; Sada, E.; Jaime, M.E.; Gonzalez, Y.; Ramachandra, L.; Rojas, R.E.; Michalak, C.; Pedraza-Sanchez, S.; Gonzalez-Noriega, A.; Torres, M. Human phagosome processing of mycobacterium tuberculosis antigens is modulated by interferon-gamma and interleukin-10. Immunology 2013, 138, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Kern, J.W.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus synthesizes adenosine to escape host immune responses. J. Exp. Med. 2009, 206, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Panther, E.; Corinti, S.; Idzko, M.; Herouy, Y.; Napp, M.; la Sala, A.; Girolomoni, G.; Norgauer, J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the t-cell stimulatory capacity of human dendritic cells. Blood 2003, 101, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Panther, E.; Idzko, M.; Herouy, Y.; Rheinen, H.; Gebicke-Haerter, P.J.; Mrowietz, U.; Dichmann, S.; Norgauer, J. Expression and function of adenosine receptors in human dendritic cells. FASEB J. 2001, 15, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by ahr and hif1-alpha. Nat. Med. 2015, 21, 638–646. [Google Scholar] [PubMed]
- Koppelman, B.; Neefjes, J.J.; de Vries, J.E.; de Waal Malefyt, R. Interleukin-10 down-regulates mhc class ii alphabeta peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity 1997, 7, 861–871. [Google Scholar] [CrossRef]
- Ding, L.; Linsley, P.S.; Huang, L.Y.; Germain, R.N.; Shevach, E.M. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 1993, 151, 1224–1234. [Google Scholar] [PubMed]
- De Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; de Vries, J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Foster, L.A.; Kurimoto, M.; Okamura, H.; Nakamura, R.M.; Kawajiri, K.; Justice, J.P.; van Scott, M.R.; Myrvik, Q.N.; Metzger, W.J. Immunoregulatory roles of IL-10 in innate immunity: IL-10 inhibits macrophage production of ifn-gamma-inducing factors but enhances nk cell production of ifn-gamma. J. Immunol. 1998, 161, 4283–4288. [Google Scholar] [PubMed]
- Nagamatsu, K.; Kuwae, A.; Konaka, T.; Nagai, S.; Yoshida, S.; Eguchi, M.; Watanabe, M.; Mimuro, H.; Koyasu, S.; Abe, A. Bordetella evades the host immune system by inducing IL-10 through a type iii effector, bopn. J. Exp. Med. 2009, 206, 3073–3088. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Nicolaos, N.; Lucet, J.C.; Daubie, C.; Benchaba, F.; Rajguru, M.; Ruimy, R.; Andremont, A.; Armand-Lefevre, L. Maternal vaginal colonisation by Staphylococcus aureus and newborn acquisition at delivery. Paediatr. Perinat Epidemiol. 2010, 24, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Shinefield, H.; Black, S.; Fattom, A.; Horwith, G.; Rasgon, S.; Ordonez, J.; Yeoh, H.; Law, D.; Robbins, J.B.; Schneerson, R.; et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N. Engl. J. Med. 2002, 346, 491–496. [Google Scholar] [PubMed]
- DeJonge, M.; Burchfield, D.; Bloom, B.; Duenas, M.; Walker, W.; Polak, M.; Jung, E.; Millard, D.; Schelonka, R.; Eyal, F.; et al. Clinical trial of safety and efficacy of INH-A21 for the prevention of nosocomial staphylococcal bloodstream infection in premature infants. J. Pediatr. 2007, 151, 260–265. [Google Scholar] [PubMed]
- Pauli, N.T.; Kim, H.K.; Falugi, F.; Huang, M.; Dulac, J.; Henry Dunand, C.; Zheng, N.Y.; Kaur, K.; Andrews, S.F.; Huang, Y.; et al. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J. Exp. Med. 2014, 211, 2331–2339. [Google Scholar] [PubMed]
- Bean, A.G.; Freiberg, R.A.; Andrade, S.; Menon, S.; Zlotnik, A. Interleukin 10 protects mice against staphylococcal enterotoxin b-induced lethal shock. Infect. Immun. 1993, 61, 4937–4939. [Google Scholar] [PubMed]
- Krakauer, T. Inhibition of toxic shock syndrome toxin-1-induced cytokine production and T cell activation by interleukin-10, interleukin-4, and dexamethasone. J. Infect. Dis. 1995, 172, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Marlow, G.J.; van Gent, D.; Ferguson, L.R. Why interleukin-10 supplementation does not work in Crohn's disease patients. World J. Gastroenterol. 2013, 19, 3931–3941. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in cancer: Any good news? Front. Oncol. 2013, 3, 108. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Roda, D.; Geuna, E.; Jimenez, B.; Rihawi, K.; Capelan, M.; Yap, T.A.; Molife, L.R.; Kaye, S.B.; de Bono, J.S.; et al. Higher risk of infections with PI3K-Akt-mTOR pathway inhibitors in patients with advanced solid tumors on phase i clinical trials. Clin. Cancer Res. 2015, 21, 1869–1876. [Google Scholar] [PubMed]
- Kaymakcalan, M.D.; Je, Y.; Sonpavde, G.; Galsky, M.; Nguyen, P.L.; Heng, D.Y.; Richards, C.J.; Choueiri, T.K. Risk of infections in renal cell carcinoma (RCC) and non-RCC patients treated with mammalian target of rapamycin inhibitors. Br. J. Cancer 2013, 108, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009, 21, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.M.; Kuglstatter, A.; Lou, Y.; Soth, M.J. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J. Med. Chem. 2010, 53, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Cohen, S.B.; Wofsy, D.; Weinblatt, M.E.; Firestein, G.S.; Brahn, E.; Strand, V.; Baker, D.G.; Tong, S.E. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral scio-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J. Rheumatol. 2011, 38, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Cheng, T.T.; Chindalore, V.; Damjanov, N.; Burgos-Vargas, R.; Delora, P.; Zimany, K.; Travers, H.; Caulfield, J.P. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009, 60, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Damjanov, N.; Kauffman, R.S.; Spencer-Green, G.T. Efficacy, pharmacodynamics, and safety of vx-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: Results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 2009, 60, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Gibbons, A.E.; Bergstrom, R.; Winn, V. The eagle effect revisited: Efficacy of clindamycin, erythromycin, and penicillin in the treatment of streptococcal myositis. J. Infect. Dis. 1988, 158, 23–28. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Peres, A.G.; Damian, A.C.; Madrenas, J. Immunomodulation and Disease Tolerance to Staphylococcus aureus. Pathogens 2015, 4, 793-815. https://doi.org/10.3390/pathogens4040793
Li Z, Peres AG, Damian AC, Madrenas J. Immunomodulation and Disease Tolerance to Staphylococcus aureus. Pathogens. 2015; 4(4):793-815. https://doi.org/10.3390/pathogens4040793
Chicago/Turabian StyleLi, Zhigang, Adam G. Peres, Andreea C. Damian, and Joaquín Madrenas. 2015. "Immunomodulation and Disease Tolerance to Staphylococcus aureus" Pathogens 4, no. 4: 793-815. https://doi.org/10.3390/pathogens4040793
APA StyleLi, Z., Peres, A. G., Damian, A. C., & Madrenas, J. (2015). Immunomodulation and Disease Tolerance to Staphylococcus aureus. Pathogens, 4(4), 793-815. https://doi.org/10.3390/pathogens4040793