
Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus


Abstract
:1. Introduction
2. The Macrophage: A Sentinel of Immunity
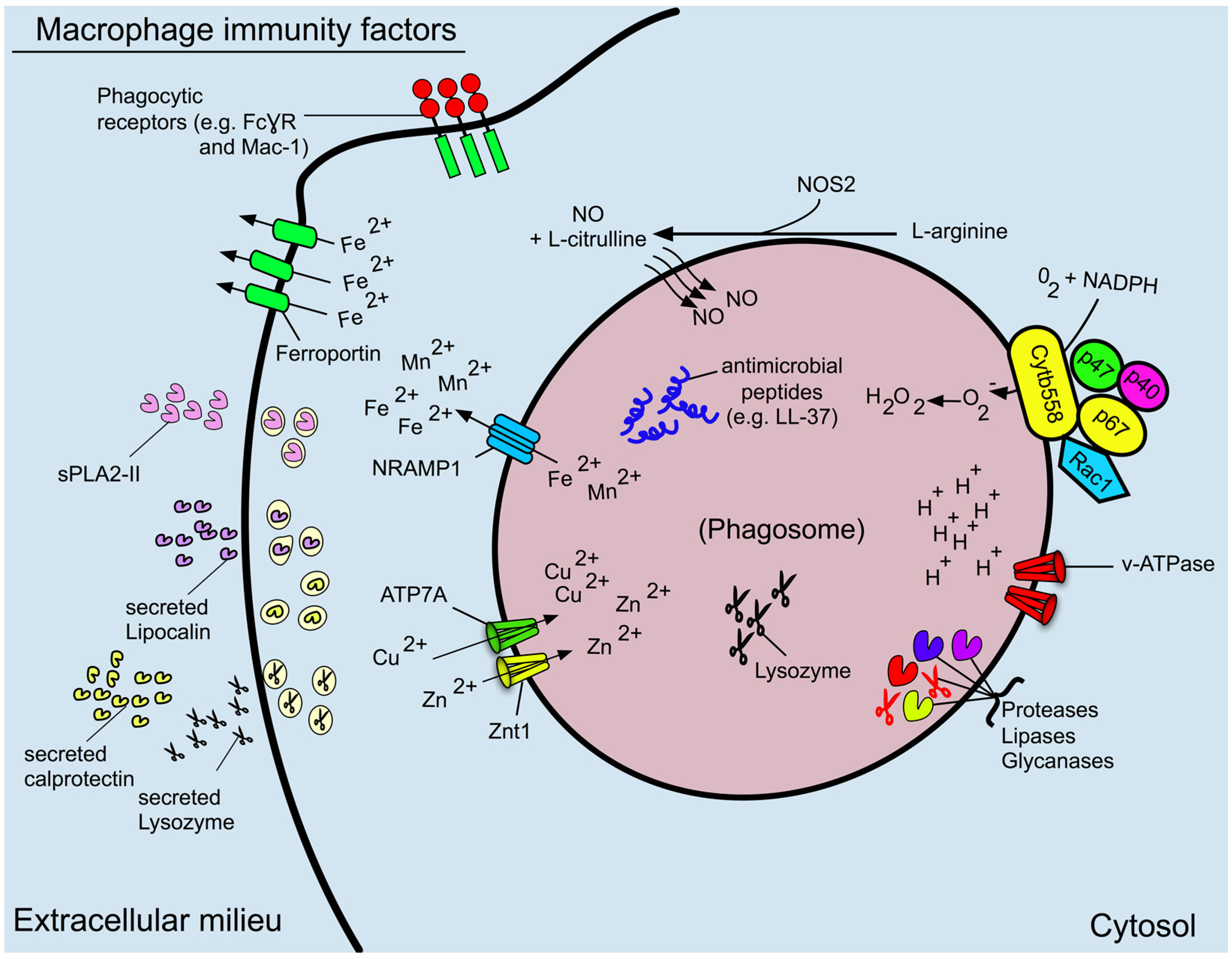
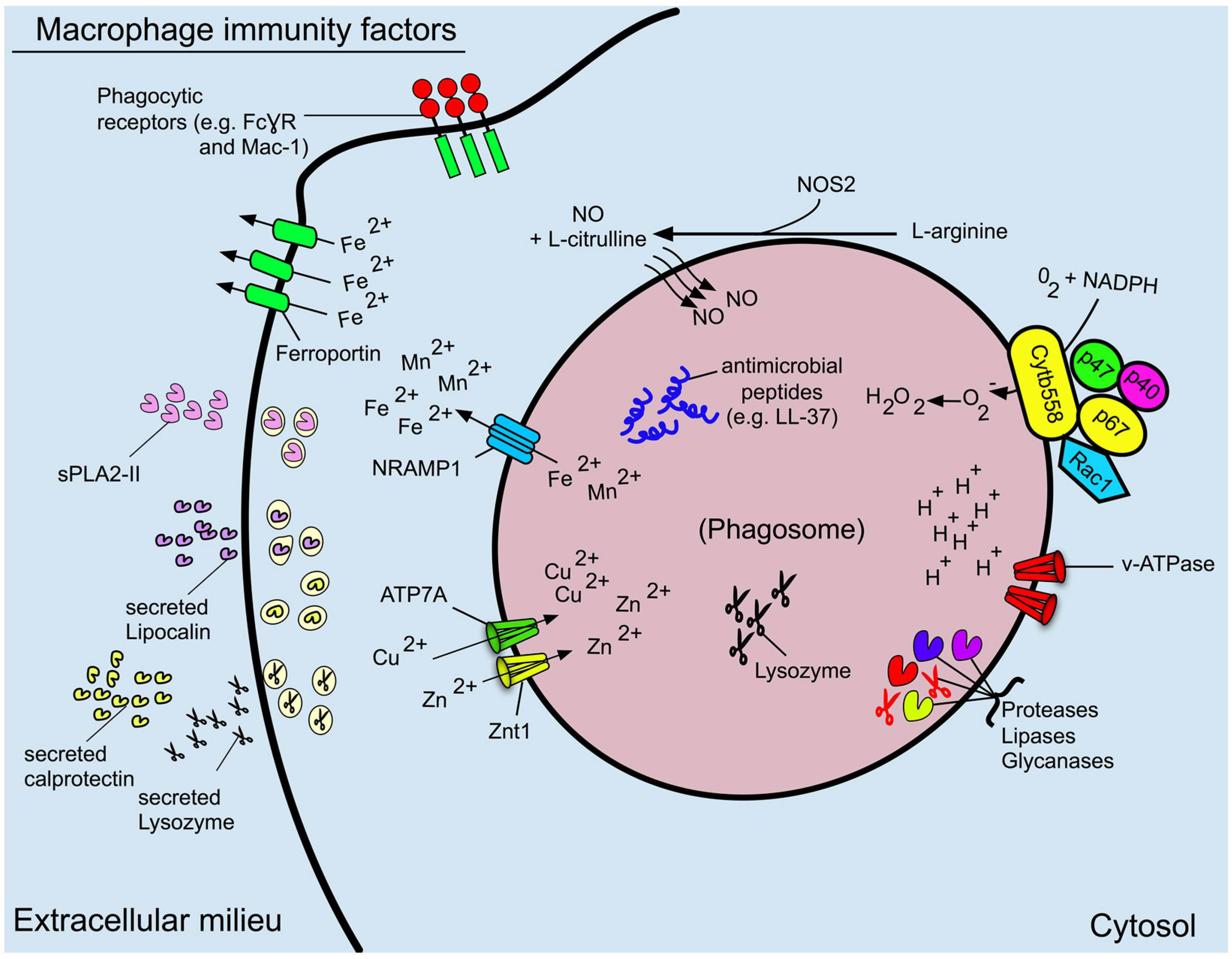
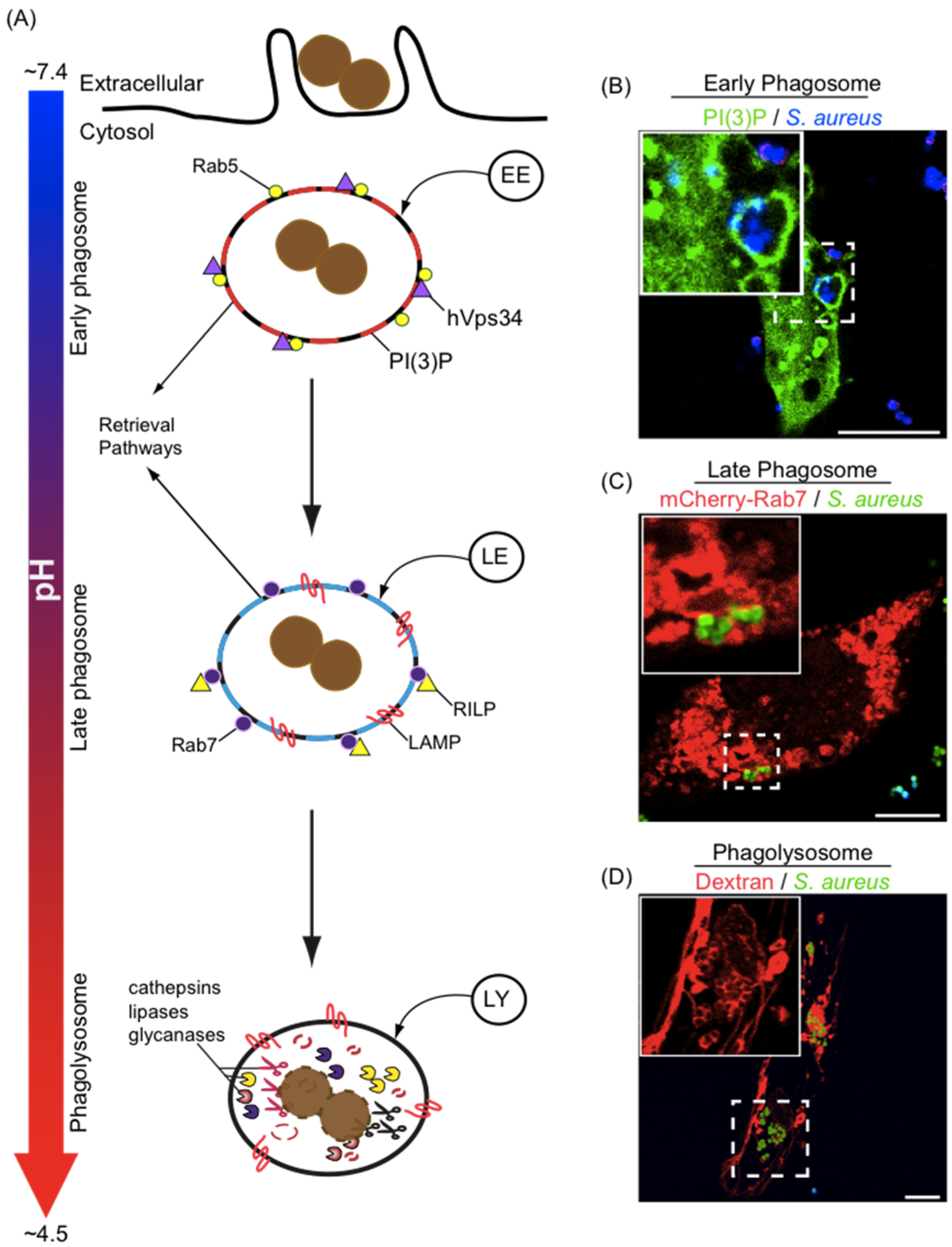
3. Phagosome Formation and Maturation
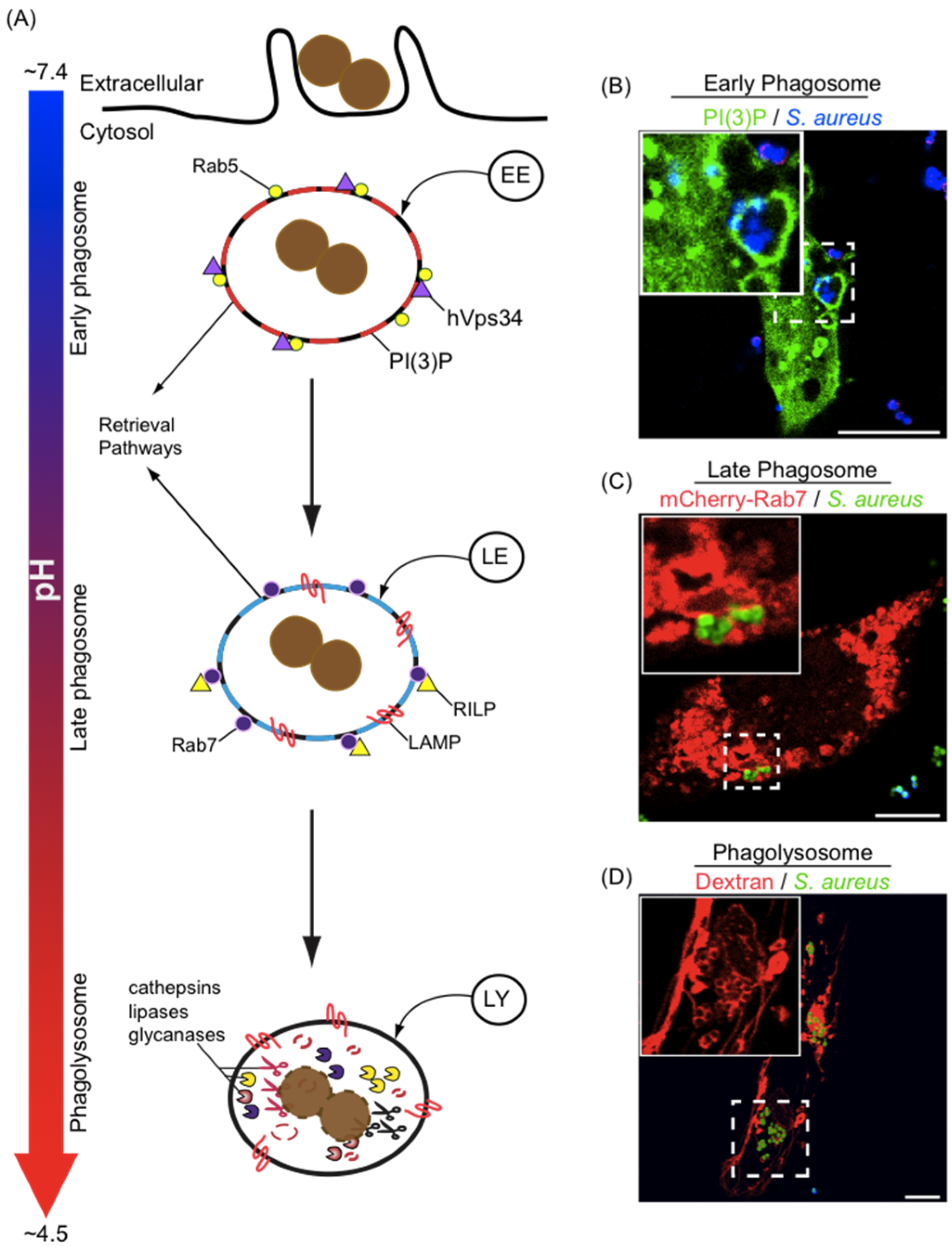
4. Phagosome Acidification
5. Reactive Oxygen and Nitrogen Species
6. Antimicrobial Proteins and Peptides
7. The Role of the Macrophage in Nutritional Immunity
8. Macrophage Extracellular Traps (mETs)
9. S. aureus Evasion of Macrophage Defenses
10. Extracellular Intoxication of Phagocytes

{kind=link}
{kind=link}
| Evasion Strategy | Factor | Description | Ref. |
|---|---|---|---|
| Host cell intoxication | Leukotoxins | ||
| LukAB | Pore forming toxin; S subunit LukA engages CD11b subunit of Mac-1; targets macrophages and neutrophils of human origin | [167] | |
| LukED | Pore forming toxin; S subunit LukE engages CCR5, CXCR1/2, and DARC; targets macrophages, neutrophils, T-lymphocytes and red blood cells from many animal species | [164,168,169] | |
| LukSF-PV | Pore forming toxin; S subunit LukS engages complement receptors C5aR and C5aR2 of human and rabbit origin, targets neutrophils, monocytes and macrophages | [170,171] | |
| HlgAB | Pore forming toxin; S subunit HlgA engages CXCR1, CXCR2 and CCR2; targets neutrophils, monocytes and macrophages of human and murine origin with the exception that murine neutrophils are resistant to lysis | [172] | |
| Host cell intoxication | Leukotoxins | ||
| HlgCB | Pore forming toxin, S subunit HlgC engages C5aR1 and C5aR2 to target neutrophils, monocytes and macrophages; demonstrates broad species specificity excluding mouse | [171,172] | |
| α-hemolysisn | Pore forming toxin; Utilizes host protein ADAM10 as receptor; Targets many cell types including macrophages of many origins including mice and humans | [173,174] | |
| α-PSMs | Small amphipathic peptides; broad lytic activity in vitro; may function as intracellular lysins | [175,176] | |
| Avoidance of Phagocytosis | Opsonin Interference | ||
| Protein A and Sbi | Bind Fc region of IgG, occlude Fc region to prevent FcγR and C1q recognition | [177,178] | |
| Staphylokinase | Bacterial plasminogen activator; activates serine protease plasmin to promote degradation of complement and Ig | [179] | |
| Aureolysin | Secreted metalloprotease; degrades complement to prevent C3b opsonization | [180] | |
| Staphopain A/B | Secreted cysteine proteases; degrade complement thereby preventing opsonization | [181] | |
| V8 | Secreted serine protease; degrades complement components and IgG | [181] | |
| Efb | Secreted bi-functional fibrinogen and C3b binding protein; Masks C3b on bacterial surface by promoting formation of a fibrinogen “shield” | [182] | |
| Capsule polysaccharide | Secreted polysaccharide polymer that encases the bacteria; shields bacterial surface from opsonins | [183,184] | |
| Complement inhibition | |||
| Cna | A collagen binding surface expressed protein; binds complement protein C1q; blocks C1q-dependent complement activation | [185] | |
| SCIN | Small secreted molecule; binds directly C3 convertase required for processing of C3 to C3a and C3b to inhibit convertase function | [186] | |
| Sbi | A cell wall associated and secreted protein; can recruit human plasminogen that is converted to plasmin to degrade C3; Can bind C3 products with the complement regulatory factor H to promote Factor I cleavage of C3b to inactive iC3b | [187,188] | |
| SdrE | Cell surface associated protein; binds Factor H recruiting it to the bacterial surface where it can act as a co-factor with Factor I to promote cleavage of C3b to iC3b | [189] | |
| ClfA | Cell-wall associated fibrinogen binding protein; binds Factor I that mediates cleavage of C3b to its inactive form iC3b | [190,191] | |
| Evasion of macrophage anti-microbial defenses | Bacterial cell surface modification | ||
| OatA | Acetylates peptidoglycan to confer resistance to lysozyme | [192] | |
| DltABCD | Catalyze the incorporation of D-alanine into wall teichoic acids to reduce the negative charge of the bacterial cell surface; decreases binding of cationic antimicrobial peptides | [193] | |
| MprF | Catalyzes modification of negatively charged cytoplasmic membrane lipids by incorporating lysine residues to make the membrane less anionic; decreases binding of cationic antimicrobial peptides | [194] | |
| Eap, EapH1, EapH2 | Secreted proteins that selectively inhibit the serine proteases neutrophil elastase, proteinase 3, and cathepsin G that are expressed by neutrophils; may function to inhibit serine proteases expressed by macrophages | [195] | |
| Resistance to ROS and RNS | |||
| SodA and SodM | Superoxide dismutases that detoxify ROS by catalyzing the conversion of superoxide into hydrogen peroxide | [196] | |
| KatA | A catalase that detoxifies hydrogen peroxide by catalyzing its breakdown into water and oxygen | [197] | |
| Msr | Methionine sulfoxide reductase; catalyzes the repair of methionine residues damaged by oxidation. | [198] | |
| Staphyloxanthin | A carotenoid expressed by S. aureus giving the cocci its golden pigmentation but can also, because of its molecular structure, act as an antioxidant; May also promote resistance to antimicrobial peptides as staphyloxanthin production decreases membrane fluidity | [199,200] | |
| Ldh1 | A S. aureus specific lactate dehydrogenase that catalyzes the reduction of pyruvate to l-lactate with the concomitant oxidation of NADH to NAD+; helps maintain cellular redox balance in cells that are unable to respire because of nitric oxide mediated damage to electron transport chain proteins | [201] | |
| Hmp | Flavohemoprotein that scavenges NO to minimize damage to other S. aureus cellular components; up-regulated in response to nitrosative stress | [202,203] | |
| Overcoming nutritional immunity | Divalent metal acquisition systems | ||
| Staphyloferrin A | Citrate based siderophore of S. aureus; extracts iron from host proteins to support staphylococcal growth; does not bind lipocalin | [204] | |
| Staphyloferrin B | Citrate based siderophore of S. aureus; extracts iron from host proteins to support staphylococcal growth; does not bind lipocalin | [205] | |
| Sst | Staphylococcal siderophore transport locus; encodes ABC transporter required for utilization of catechol siderophores and host derived, iron-binding stress hormones (e.g., norepinephrine) | [206] | |
| Fhu | The fhuD1/2 and fhuCBG genes encode the receptors and permease, respectively, needed for utilization of hydroxymate type siderophores | [207,208] | |
| Isd | Isd proteins collectively allow S. aureus to bind hemoglobin, remove heme and shuttle it to the cytoplasm for use as an iron source | [209] | |
| MntABC and MntH | Transporters of Mn2+, required for growth under Mn2+ limited conditions (e.g., inside abscesses) | [210] |
11. Evasion of Complement and Opsono-Phagocytosis
12. Evasion of Opsono-Phagocytosis
13. S. aureus Inhibition of Complement Activation
14. Resistance to Phagolysosomal Killing and Host Antimicrobial Proteins
15. S. aureus Resistance to Oxidative and Nitrosative Killing
16. Overcoming Nutritional Immunity
17. Concluding Remarks
Acknowledgments
Author Contributions
Conflict of Interest
References
- Van Hal, S.J.; Jensen, S.O.; Vaska, V.L.; Espedido, B.A.; Paterson, D.L.; Gosbell, I.B. Predictors of mortality in Staphylococcus aureus bacteremia. Clin. Microbiol. Rev. 2012, 25, 362–386. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, K.D.; Xu, J.; Murphy, S.L.; Miniño, A.M.; Kung, H.-C. Deaths: Final data for 2009. Natl. Vital Stat. Reports 2012, 60, 1–116. [Google Scholar]
- Boucher, H.; Miller, L.G.; Razonable, R.R. Serious infections caused by methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2010, 51 Suppl 2, S183–197. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, J.R.; Chen, L.; Mathema, B.; Kreiswirth, B.N. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA). Curr. Opin. Microbiol. 2012, 15, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, G.R. USA300 abroad: Global spread of a virulent strain of community-associated methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2012, 18, 725–34. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, L.R.; Joshi, G.S.; Richardson, A.R. Virulence strategies of the dominant USA300 lineage of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol Med Microbiol 2012, 65, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, B.E.; Martinez-Aguilar, G.; Hulten, K.G.; Hammerman, W.A.; Coss-Bu, J.; Avalos-Mishaan, A.; Mason, E.O.J.; Kaplan, S.L. Severe Staphylococcal sepsis in adolescents in the era of community-acquired methicillin-resistant Staphylococcus aureus. Pediatrics 2005, 115, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Rey, M. Staphylococcus aureus Panton Valentine leukocidin causes necrotizing pneumonia. Science 2007, 315, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Saavedra-Lozano, J.; Mejías, A.; Ahmad, N.; Peromingo, E.; Ardura, M.I.; Guillen, S.; Syed, A.; Cavuoti, D.; Ramilo, O. Changing trends in acute osteomyelitis in children: Impact of methicillin-resistant Staphylococcus aureus infections. J. Pediatr. Orthop. 2015, 28, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Surewaard, B.G.J.; Nijland, R.; van Strijp, J.A.G. Neutrophils versus Staphylococcus aureus: A biological tug of war. Annu. Rev. Microbiol. 2013, 67, 629–50. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Sharma, M.; Ung, C.; Tuli, A.; Barral, D.C.; Hava, D.L.; Veerapen, N.; Besra, G.S.; Hacohen, N.; Brenner, M.B. Lysosomal Trafficking, Antigen Presentation, and Microbial Killing Are Controlled by the Arf-like GTPase Arl8b. Immunity 2011, 35, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–7. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; Rodewald, H.-R. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2014, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol 2012, 188, 5752–5765. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Huang, C.; Lin, Z.; Zhan, S.; Kong, L.; Fang, C.; Li, J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell. Signal. 2014, 26, 192–7. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Araki, N.; Johnson, M.T.; Swanson, J.A. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 1996, 135, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Schlam, D.; Bagshaw, R.D.; Freeman, S.A.; Collins, R.F.; Pawson, T.; Fairn, G.D.; Grinstein, S. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through Rac/Cdc42 GTPase-activating proteins. Nat. Commun. 2015, 6, 8623. [Google Scholar] [CrossRef] [PubMed]
- Pitt, A.; Mayorga, L.S.; Stahl, P.D.; Schwartz, A.L. Alterations in the protein composition of maturing phagosomes. J. Clin. Invest. 1992, 90, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Hackam, D.J.; Rotstein, O.D.; Zhang, W.J.; Demaurex, N.; Woodside, M.; Tsai, O.; Grinstein, S. Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K+-ATPases, and vacuolar-type H+-atpases. J Biol Chem 1997, 272, 29810–29820. [Google Scholar] [CrossRef] [PubMed]
- Fairn, G.D.; Grinstein, S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. 2012, 33, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Huber, L.A.; Parton, R.G.; Griffiths, G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 1994, 124, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell. Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Herre, J.; Marshall, A.S.J.; Caron, E.; Edwards, A.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.; Sousa, C.R.E.; Gordon, S.; Brown, G.D. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004, 104, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.C.; Harrison, R.E. Membrane ruffles capture C3bi-opsonized particles in activated macrophages. Mol. Biol. Cell 2008, 19, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Downey, G.P.; Botelho, R.J.; Butler, J.R.; Moltyaner, Y.; Chien, P.; Schreiber, A.D.; Grinstein, S. Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcgammaRIIA receptors. J. Biol. Chem. 1999, 274, 28436–28444. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Harrison, R.E.; Yip, C.M.; Jaqaman, K.; Grinstein, S. Dynamic macrophage “probing” is required for the efficient capture of phagocytic targets. J. Cell Biol. 2010, 191, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Bohdanowicz, M.; Schlam, D.; Hermansson, M.; Rizzuti, D.; Fairn, G.D.; Ueyama, T.; Somerharju, P.; Du, G.; Grinstein, S. Phosphatidic acid is required for the constitutive ruffling and macropinocytosis of phagocytes. Mol. Biol. Cell 2013, 24, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; Grinstein, S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Jaumouillé, V.; Farkash, Y.; Jaqaman, K.; Das, R.; Lowell, C.A.; Grinstein, S. Actin cytoskeleton reorganization by Syk regulates Fcγ receptor responsiveness by increasing its lateral mobility and clustering. Dev. Cell 2014, 29, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.; el Khoury, J.; di Virgilio, F.; Kaplan, E.M.; Silverstein, S.C. Ca(2+)-independent F-actin assembly and disassembly during Fc receptor-mediated phagocytosis in mouse macrophages. J. Cell Biol. 1991, 113, 757–67. [Google Scholar] [CrossRef] [PubMed]
- Tse, S.M.L.; Furuya, W.; Gold, E.; Schreiber, A.D.; Sandvig, K.; Inman, R.D.; Grinstein, S. Differential role of actin, clathrin, and dynamin in Fc gamma receptor-mediated endocytosis and phagocytosis. J. Biol. Chem. 2003, 278, 3331–8. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.C.; Dobson, W.; Botelho, R.J.; Coady-Osberg, N.; Chavrier, P.; Knecht, D.A.; Heath, C.; Stahl, P.; Grinstein, S. Phosphatidylinositol-4,5-bisphosphate hydrolysis directs actin remodeling during phagocytosis. J. Cell Biol. 2005, 169, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Fratti, R.A.; Backer, J.M.; Gruenberg, J.; Corvera, S.; Deretic, V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J. Cell Biol. 2001, 154, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Duclos, S.; Diez, R.; Garin, J.; Papadopoulou, B.; Descoteaux, A.; Stenmark, H.; Desjardins, M. Rab5 regulates the kiss and run fusion between phagosomes and endosomes and the acquisition of phagosome leishmanicidal properties in RAW 264.7 macrophages. J. Cell Sci. 2000, 113 Pt 19, 3531–3541. [Google Scholar] [PubMed]
- Shin, H.-W.; Hayashi, M.; Christoforidis, S.; Lacas-Gervais, S.; Hoepfner, S.; Wenk, M.R.; Modregger, J.; Uttenweiler-Joseph, S.; Wilm, M.; Nystuen, A.; et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J. Cell Biol. 2005, 170, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Botelho, R.J.; Rameh, L.; Brachmann, S.M.; Matsuo, T.; Davidson, H.W.; Schreiber, A.; Backer, J.M.; Cantley, L.C.; Grinstein, S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 2001, 155, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the Switch in Early-to-Late Endosome Transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.K.; Plumb, J.D.; Downey, G.P.; Valvano, M.A.; Grinstein, S. Inactivation of macrophage Rab7 by Burkholderia cenocepacia. J. Innate Immun. 2010, 2, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.E.; Bucci, C.; Vieira, O. V; Schroer, T.A.; Grinstein, S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: Role of Rab7 and RILP. Mol. Cell. Biol. 2003, 23, 6494–6506. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. CB 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.K.; Eskelinen, E.-L.; Scott, C.C.; Malevanets, A.; Saftig, P.; Grinstein, S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Damiani, M.T.; Pavarotti, M.; Leiva, N.; Lindsay, A.J.; McCaffrey, M.W.; Colombo, M.I. Rab coupling protein associates with phagosomes and regulates recycling from the phagosomal compartment. Traffic 2004, 5, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Lee, D.J.; Dale, B.M.; Calafat, J.; Greenberg, S. A Rab11-containing rapidly recycling compartment in macrophages that promotes phagocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xiao, H.; Zhang, K.; Wang, B.; Gao, Z.; Jian, Y.; Qi, X.; Sun, J.; Miao, L.; Yang, C. Retromer is required for apoptotic cell clearance by phagocytic receptor recycling. Science 2010, 327, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Booth, J.W. Divergent intracellular sorting of FcγRIIA and FcγRIIB2. J. Biol. Chem. 2010, 285, 34250–34258. [Google Scholar] [CrossRef] [PubMed]
- Schröder, B.A.; Wrocklage, C.; Hasilik, A.; Saftig, P. The proteome of lysosomes. Proteomics 2010, 10, 4053–4076. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lamothe, J.; Huynh, K.K.; Grinstein, S.; Valvano, M.A. Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell. Microbiol. 2007, 9, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Cano, V.; March, C.; Insua, J.L.; Aguiló, N.; Llobet, E.; Moranta, D.; Regueiro, V.; Brennan, G.P.; Millán-Lou, M.I.; Martín, C.; et al. Klebsiella pneumoniae survives within macrophages by avoiding delivery to lysosomes. Cell. Microbiol. 2015, 44, 1537–1560. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Swenson, D.L.; Alonso, S.; Anderson, J.; Wang, C.; Russell, D.G. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc. Natl. Acad. Sci. USA 2004, 101, 13642–13647. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, G.L.; Rotstein, O.D.; Grinstein, S. Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)-ATPase activity, counterion conductance, and H+ “leak.”. J. Biol. Chem. 1991, 266, 24540–24548. [Google Scholar] [PubMed]
- Tougard, C.; Louvard, D.; Picart, R.; Tixier-Vidal, A. Antibodies against a lysosomal membrane antigen recognize a prelysosomal compartment involved in the endocytic pathway in cultured prolactin cells. J. Cell Biol. 1985, 100, 786–793. [Google Scholar] [CrossRef] [PubMed]
- DiCiccio, J.E.; Steinberg, B.E. Lysosomal pH and analysis of the counter ion pathways that support acidification. J. Gen. Physiol. 2011, 137, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Yang, B.; Sonawane, N.D.; Sasaki, S.; Uchida, S.; Verkman, A.S. ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J. Biol. Chem. 2005, 280, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Salao, K.; Li, H.; Rybicka, J.M.; Yates, R.M.; Luo, X.W.; Shi, X.X.; Kuffner, T.; Tsai, V.W.-W.; Husaini, Y.; et al. Intracellular chloride channel protein CLIC1 regulates macrophage function through modulation of phagosomal acidification. J. Cell Sci. 2012, 125, 5479–5488. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Sokolovska, A.; Charriere, G.M.; Boyer, L.; Dejardin, S.; Cappillino, M.P.; Yantosca, L.M.; Takahashi, K.; Moore, K.J.; Lacy-Hulbert, A.; et al. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J. Immunol. 2010, 184, 7071–7081. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.H.; Hart, P.D.; Young, M.R. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature 1980, 286, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Dolenc, I.; Turk, V.; Bieth, J.G. Kinetics of the pH-induced inactivation of human cathepsin L. Biochemistry 1993, 32, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Jabado, N.; Jankowski, A.; Dougaparsad, S.; Picard, V.; Grinstein, S.; Gros, P. Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J. Exp. Med. 2000, 192, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, D. Recent advances in chronic granulomatous disease. J. Infect. 2014, 69, S32–S35. [Google Scholar] [CrossRef] [PubMed]
- Lam, G.Y.; Huang, J.; Brumell, J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol. 2010, 32, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Price, M.O.; Atkinson, S.J.; Knaus, U.G.; Dinauer, M.C. Rac activation induces NADPH oxidase activity in transgenic COSphox cells, and the level of superoxide production is exchange factor-dependent. J. Biol. Chem. 2002, 277, 19220–19228. [Google Scholar] [CrossRef] [PubMed]
- Price, M.O.; McPhail, L.C.; Lambeth, J.D.; Han, C.H.; Knaus, U.G.; Dinauer, M.C. Creation of a genetic system for analysis of the phagocyte respiratory burst: High-level reconstitution of the NADPH oxidase in a nonhematopoietic system. Blood 2002, 99, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Rotrosen, D.; Yeung, C.L.; Leto, T.L.; Malech, H.L.; Kwong, C.H. Cytochrome b558: The flavin-binding component of the phagocyte NADPH oxidase. Science 1992, 256, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–70. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G.; Heyworth, P.G.; Evans, T.; Curnutte, J.T.; Bokoch, G.M. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 1991, 254, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Chessa, T.A.M.; Davidson, K.; Henderson, R.B.; Walker, S.; Tolmachova, T.; Grys, K.; Rausch, O.; Seabra, M.C.; Tybulewicz, V.L.J.; et al. PtdIns3P and Rac direct the assembly of the NADPH oxidase on a novel, pre-phagosomal compartment during FcR-mediated phagocytosis in primary mouse neutrophils. Blood 2010, 116, 4978–4989. [Google Scholar] [CrossRef] [PubMed]
- Schlam, D.; Bohdanowicz, M.; Chatilialoglu, A.; Steinberg, B.E.; Ueyama, T.; Du, G.; Grinstein, S.; Fairn, G.D. Diacylglycerol Kinases Terminate Diacylglycerol Signaling during the Respiratory Burst Leading to Heterogeneous Phagosomal NADPH Oxidase Activation. J. Biol. Chem. 2013, 288, 23090–23104. [Google Scholar] [CrossRef] [PubMed]
- Casbon, A.-J.; Allen, L.-A.H.; Dunn, K.W.; Dinauer, M.C. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J. Immunol. 2009, 182, 2325–2339. [Google Scholar] [CrossRef] [PubMed]
- Balce, D.R.; Li, B.; Allan, E.R.O.; Rybicka, J.M.; Krohn, R.M.; Yates, R.M. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood 2011, 118, 4199–4208. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.-M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed]
- El Chemaly, A.; Nunes, P.; Jimaja, W.; Castelbou, C.; Demaurex, N. Hv1 proton channels differentially regulate the pH of neutrophil and macrophage phagosomes by sustaining the production of phagosomal ROS that inhibit the delivery of vacuolar ATPases. J. Leukoc. Biol. 2014, 95, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C.; Röllinghoff, M.; Diefenbach, A. The role of nitric oxide in innate immunity. Immunol. Rev. 2000, 173, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Misukonis, M.A.; Shami, P.J.; Mason, S.N.; Sauls, D.L.; Dittman, W.A.; Wood, E.R.; Smith, G.K.; McDonald, B.; Bachus, K.E. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 1995, 86, 1184–1195. [Google Scholar] [PubMed]
- Schneemann, M.; Schoeden, G. Macrophage biology and immunology: man is not a mouse. J. Leukoc. Biol. 2007, 81, 579, discussion 580. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C.; Nathan, C.F. Man is not a mouse: reply. J. Leukoc. Biol. 2007, 81, 580. [Google Scholar] [CrossRef] [PubMed]
- Denis, M. Tumor necrosis factor and granulocyte macrophage-colony stimulating factor stimulate human macrophages to restrict growth of virulent Mycobacterium avium and to kill avirulent M. avium: killing effector mechanism depends on the generation of. J. Leukoc. Biol. 1991, 49, 380–7. [Google Scholar] [PubMed]
- Bermudez, L.E. Differential mechanisms of intracellular killing of Mycobacterium avium and Listeria monocytogenes by activated human and murine macrophages. The role of nitric oxide. Clin. Exp. Immunol. 1993, 91, 277–81. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Harvey, M.W.; Holden, D.W.; Evans, T.J. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect. Immun. 2001, 69, 6391–6400. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.S.; Vergne, I.; Master, S.S.; Kyei, G.B.; Chua, J.; Deretic, V. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLoS Pathog. 2007, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.H.; Fratti, R.A.; Poschet, J.F.; Timmins, G.S.; Master, S.S.; Burgos, M.; Marletta, M.A.; Deretic, V. Mycobacteria Inhibit Nitric Oxide Synthase Recruitment to Phagosomes during Macrophage Infection. Infect. Immun. 2004, 72, 2872–2878. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Russell, D.; Xie, Q.W.; Bogdan, C.; Nathan, C. Vesicle membrane association of nitric oxide synthase in primary mouse macrophages. J. Immunol. 1995, 154, 2914–2925. [Google Scholar] [PubMed]
- Richardson, A.R.; Soliven, K.C.; Castor, M.E.; Barnes, P.D.; Libby, S.J.; Fang, F.C. The Base Excision Repair system of Salmonella enterica serovar Typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog. 2009, 5, e1000451. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.R.; Payne, E.C.; Younger, N.; Karlinsey, J.E.; Thomas, V.C.; Becker, L.A.; Navarre, W.W.; Castor, M.E.; Libby, S.J.; Fang, F.C. Multiple targets of nitric oxide in the tricarboxylic acid cycle of Salmonella enterica serovar Typhimurium. Cell Host Microbe 2011, 10, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Akaike, T.; Okamoto, S.; Kubota, T.; Yoshitake, J.; Sawa, T.; Miyamoto, Y.; Tamura, F.; Maeda, H. Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect. Immun. 2002, 70, 3130–42. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Schleicher, U.; Schroll, A.; Sonnweber, T.; Theurl, I.; Ludwiczek, S.; Talasz, H.; Brandacher, G.; Moser, P.L.; Muckenthaler, M.U.; et al. Nitric oxide–mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J. Exp. Med. 2013, 275, 19906. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Cosío, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Faulhaber, A.; Sieber, C.; Pfeifer, D.; Hochberg, T.; Gansz, M.; Deshmukh, S.D.; Dauth, S.; Brix, K.; Saftig, P.; et al. The endolysosomal cysteine cathepsins L and K are involved in macrophage-mediated clearance of Staphylococcus aureus and the concomitant cytokine induction. FASEB J. 2014, 28, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Del Cerro-Vadillo, E.; Madrazo-Toca, F.; Carrasco-Marin, E.; Fernandez-Prieto, L.; Beck, C.; Leyva-Cobian, F.; Saftig, P.; Alvarez-Dominguez, C. Cutting edge: A novel nonoxidative phagosomal mechanism exerted by cathepsin-D controls Listeria monocytogenes intracellular growth. J. Immunol. 2006, 176, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro-IL-1 Synthesis and NLRP3-Mediated IL-1 Activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, J.; Sasadaira, H.; Watanabe, K.; Watanabe, Y. Ultrastructural immunocytochemical localization of lysozyme in human monocytes and macrophages. Cell Tissue Res. 1985, 242, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.; Karunakaran, D.; Traini, M.; Xue, J.; Huang, K.Y.; Nawara, D.; Gaus, K.; Jessup, W.; Robinson, P.J.; Kritharides, L. Pharmacological inhibition of dynamin II reduces constitutive protein secretion from primary human macrophages. PLoS One 2014, 9, e111186. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Toibana, A.; Nakahama, K. Human lysozyme: Sequencing of a cDNA, and expression and secretion by Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1988, 150, 794–801. [Google Scholar] [CrossRef]
- Tang, X.; Basavarajappa, D.; Haeggström, J.Z.; Wan, M. P2X7 Receptor Regulates Internalization of Antimicrobial Peptide LL-37 by Human Macrophages That Promotes Intracellular Pathogen Clearance. J. Immunol. 2015, 195, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.; Santos, J.C.; Mishra, B.B.; Jena, P.; Progida, C.; Sorensen, O.E.; Gallo, R.; Appelberg, R.; Griffiths, G. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell. Microbiol. 2011, 13, 1601–1617. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Shin, D.-M.; Lee, H.-M.; Yang, C.-S.; Jin, H.S.; Kim, K.-K.; Lee, Z.-W.; Lee, S.-H.; Kim, J.-M.; Jo, E.-K. Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages via Cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Soehnlein, O.; Tang, X.; van der Does, A.M.; Smedler, E.; Uhleń, P.; Lindbom, L.; Agerberth, B.; Haeggström, J.Z. Cathelicidin LL-37 induces time-resolved release of LTB4 and TXA 2by human macrophages and triggers eicosanoid generation in vivo. FASEB J. 2014, 28, 3456–3467. [Google Scholar] [CrossRef] [PubMed]
- Flamand, L.; Tremblay, M.J.; Borgeat, P. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. J. Immunol. 2007, 178, 8036–8045. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell. Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Raymond, B.; Goossens, P.L.; Njamkepo, E.; Guiso, N.; Paya, M.; Touqui, L. Type-IIA secreted phospholipase A2 is an endogenous antibiotic-like protein of the host. Biochimie 2010, 92, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Weinrauch, Y.; Elsbach, P.; Madsen, L.M.; Foreman, A.; Weiss, J. The potent anti-Staphylococcus aureus activity of a sterile rabbit inflammatory fluid is due to a 14-kD phospholipase A2. J. Clin. Invest. 1996, 97, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Grönroos, J.O.; Laine, V.J.O.; Nevalainen, T.J. Bactericidal group IIA phospholipase A2 in serum of patients with bacterial infections. J. Infect. Dis. 2002, 185, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Weinrauch, Y.; Abad, C.; Liang, N.S.; Lowry, S.F.; Weiss, J. Mobilization of Potent Plasma Bactericidal Activity during Systemic Bacterial Challenge. J Clin Invest 1998, 102, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Femling, J.K.; Nauseef, W.M.; Weiss, J.P. Synergy between extracellular group IIA phospholipase A2 and phagocyte NADPH oxidase in digestion of phospholipids of Staphylococcus aureus ingested by human neutrophils. J. Immunol. 2005, 175, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Gruenheid, S.; Pinner, E.; Desjardins, M.; Gros, P. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 1997, 185, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Searle, S.; Bright, N.A.; Roach, T.I.; Atkinson, P.G.; Barton, C.H.; Meloen, R.H.; Blackwell, J.M. Localisation of Nramp1 in macrophages: modulation with activation and infection. J. Cell Sci. 1998, 111, 2855–2866. [Google Scholar] [PubMed]
- Van Crevel, R.; Parwati, I.; Sahiratmadja, E.; Marzuki, S.; Ottenhoff, T.H.M.; Netea, M.G.; van der Ven, A.; Nelwan, R.H.; van der Meer, J.W.; Alisjahbana, B.; et al. Infection with Mycobacterium tuberculosis Beijing genotype strains is associated with polymorphisms in SLC11A1/NRAMP1 in Indonesian patients with tuberculosis. J. Infect. Dis. 2009, 200, 1671–1674. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Abel, L.; Tooker, H.; Poon, A.; Simkin, L.; Girard, M.; Adams, G.J.; Starke, J.R.; Smith, K.C.; Graviss, E.A.; et al. Alleles of the NRAMP1 gene are risk factors for pediatric tuberculosis disease. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12183–12188. [Google Scholar] [CrossRef] [PubMed]
- Cuellar-Mata, P.; Jabado, N.; Liu, J.; Furuya, W.; Finlay, B.B.; Gros, P.; Grinstein, S. Nramp1 modifies the fusion of Salmonella typhimurium-containing vacuoles with cellular endomembranes in macrophages. J. Biol. Chem. 2002, 277, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Watt, R.K. The many faces of the octahedral ferritin protein. Biometals 2011, 24, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Delaby, C.; Pilard, N.; Gonçalves, A.S.; Beaumont, C.; Canonne-Hergaux, F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005, 106, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Jung, G.; Gabayan, V.; Valore, E.; Ruchala, P.; Gulig, P.A.; Ganz, T.; Nemeth, E.; Bulut, Y. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe 2015, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [PubMed]
- Chlosta, S.; Fishman, D.S.; Harrington, L.; Johnson, E.E.; Knutson, M.D.; Wessling-Resnick, M.; Cherayil, B.J. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect. Immun. 2006, 74, 3065–3067. [Google Scholar] [CrossRef] [PubMed]
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Blumh, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Nairz, M.; Ferring-Appel, D.; Casarrubea, D.; Sonnweber, T.; Viatte, L.; Schroll, A.; Haschka, D.; Fang, F.C.; Hentze, M.W.; Weiss, G.; et al. Iron regulatory proteins mediate host resistance to Salmonella infection. Cell Host Microbe 2015, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, G.; Nairz, M.; Libby, S.J.; Fang, F.C.; Weiss, G. Slc11a1 (Nramp1) impairs growth of Salmonella enterica serovar Typhimurium in macrophages via stimulation of lipocalin-2 expression. J. Leukoc. Biol. 2012, 92, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Stafford, S.L.; Bokil, N.J.; Achard, M.E.S.; Kapetanovic, R.; Schembri, M.A.; McEwan, A.G.; Sweet, M.J. Metal ions in macrophage antimicrobial pathways: emerging roles for zinc and copper. Biosci. Rep. 2013, 33, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Botella, H.; Peyron, P.; Levillain, F.; Poincloux, R.; Poquet, Y.; Brandli, I.; Wang, C.; Tailleux, L.; Tilleul, S.; Charrière, G.M.; et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe 2011, 10, 248–259. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Lee, J.; Kambe, T.; Fritsche, K.; Petris, M.J. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 2009, 284, 33949–33956. [Google Scholar] [CrossRef] [PubMed]
- Achard, M.E.S.; Stafford, S.L.; Bokil, N.J.; Chartres, J.; Bernhardt, P.V.; Schembri, M.A.; Sweet, M.J.; McEwan, A.G. Copper redistribution in murine macrophages in response to Salmonella infection. Biochem. J. 2012, 444, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Osman, D.; Waldron, K.J.; Denton, H.; Taylor, C.M.; Grant, A.J.; Mastroeni, P.; Robinson, N.J.; Cavet, J.S. Copper homeostasis in Salmonella is atypical and copper-CueP is a major periplasmic metal complex. J. Biol. Chem. 2010, 285, 25259–25268. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Chow, O.A.; von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; Nizet, V. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–54. [Google Scholar] [CrossRef] [PubMed]
- Von Köckritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; Simon, H.-U. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Boe, D.M.; Curtis, B.J.; Chen, M.M.; Ippolito, J.A.; Kovacs, E.J. Extracellular traps and macrophages: new roles for the versatile phagocyte. J. Leukoc. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, X.; Liao, C.; Liu, X.; Du, J.; Shi, H.; Wang, X.; Bai, X.; Peng, P.; Yu, L.; Wang, F.; Zhao, Y.; Liu, M. Escherichia coli and Candida albicans induced macrophage extracellular trap-like structures with limited microbicidal activity. PLoS One 2014, 9, e90042. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., III; Torres, V.J. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 2014, 78, 199–230. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel). 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Otto, M. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 2013, 11, 12–14. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins-critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef] [PubMed]
- Badarau, A.; Rouha, H.; Malafa, S.; Logan, D.T.; Håkansson, M.; Stulik, L.; Dolezilkova, I.; Teubenbacher, A.; Gross, K.; Maierhofer, B.; et al. Structure-function analysis of heterodimer formation, oligomerization, and receptor binding of the Staphylococcus aureus bi-component toxin LukGH. J. Biol. Chem. 2015, 290, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Kawai, Y.; Tanaka, Y.; Hirano, N.; Kaneko, J.; Tomita, N.; Ohta, M.; Kamio, Y.; Yao, M.; Tanaka, I. Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc. Natl. Acad. Sci. 2011, 108, 17314–17319. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., III; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [PubMed]
- Colin, D.A.; Mazurier, I.; Sire, S.; Finck-Barbancon, V. Interaction of the two components of leukocidin from Staphylococcus aureus with human polymorphonuclear leukocyte membranes: Sequential binding and subsequent activation. Infect. Immun. 1994, 62, 3184–3188. [Google Scholar] [PubMed]
- DuMont, A.L.; Yoong, P.; Liu, X.; Day, C.J.; Chumbler, N.M.; James, D.B.A.; Alonzo, F., III; Bode, N.J.; Borden Lacy, D.; Jennings, M.P.; Torres, V.J. Identification of a crucial residue required for Staphylococcus aureus LukAB cytotoxicity and receptor recognition. Infect. Immun. 2014, 82, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- DuMont, A.L.; Yoong, P.; Day, C.J.; Alonzo, F., III; McDonald, W.H.; Jennings, M.P.; Torres, V.J. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10794–10799. [Google Scholar] [PubMed]
- Reyes-Robles, T.; Alonzo, F., III; Kozhaya, L.; Lacy, D.B.; Unutmaz, D.; Torres, V.J. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 2013, 14, 453–459. [Google Scholar] [PubMed]
- Spaan, A.N.; Reyes-Robles, T.; Badiou, C.; Cochet, S.; Boguslawski, K.M.; Yoong, P.; Day, C.J.; de Haas, C.J.C.; van Kessel, K.P.M.; Vandenesch, F.; et al. Staphylococcus aureus targets the Duffy Antigen Receptor for Chemokines (DARC) to lyse erythrocytes. Cell Host Microbe 2015, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Henry, T.; van Rooijen, W.J.M.; Perret, M.; Badiou, C.; Aerts, P.C.; Kemmink, J.; de Haas, C.J.C.; van Kessel, K.P.M.; Vandenesch, F.; et al. The staphylococcal toxin Panton-Valentine Leukocidin targets human C5a receptors. Cell Host Microbe 2013, 13, 584–94. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Schiepers, A.; de Haas, C.J.C.; van Hooijdonk, D.D.J.J.; Badiou, C.; Contamin, H.; Vandenesch, F.; Lina, G.; Gerard, N.P.; Gerard, C.; et al. Differential interaction of the staphylococcal toxins Panton-Valentine Leukocidin and -hemolysin CB with human C5a receptors. J. Immunol. 2015, 195, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; de Haas, C.J.C.; Day, C.J.; Jennings, M.P.; et al. The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 2014, 5, 5438. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA. 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.E.N.; Berube, B.J.; Sampedro, G.R.; DeDent, A.C.; Bubeck Wardenburg, J. Tissue-specific patterning of host innate immune responses by Staphylococcus aureus α-toxin. J. Innate Immun. 2014, 6, 619–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.-H.H.L.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A.; DeLeo, F.R.; Otto, M. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 2007, 13, 1510–1514. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.J.; de Haas, C.J.C.; Vervoort, F.; Rigby, K.M.; Deleo, F.R.; Otto, M.; van Strijp, J.A.G.; Nijland, R. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell. Microbiol. 2013, 15, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Dossett, J.H.; Kronvall, G.; Williams, R.C.; Quie, P.G. Antiphagocytic effects of staphylococcal protein A. J. Immunol. 1969, 103, 1405–1410. [Google Scholar] [PubMed]
- Zhang, L.H.; Jacobsson, K.; Vasi, J.; Lindberg, M.; Frykberg, L. A second IgG-binding protein in Staphylococcus aureus. Microbiology 1998, 144, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.M.; van Wamel, W.J.B.; Ruyken, M.; van Kessel, K.P.M.; van Strijp, J.A.G. Anti-opsonic properties of staphylokinase. Microbes Infect. 2005, 7, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.J.; Ruyken, M.; Malone, C.L.; van Strijp, J.A.G.; Horswill, A.R.; Rooijakkers, S.H.M. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J. Immunol. 2011, 186, 6445–6453. [Google Scholar] [CrossRef] [PubMed]
- Kolar, S.L.; Antonio Ibarra, J.; Rivera, F.E.; Mootz, J.M.; Davenport, J.E.; Stevens, S.M.; Horswill, A.R.; Shaw, L.N. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen 2013, 2, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.-P.P.; Kuipers, A.; Freitag, C.M.; Jongerius, I.; Medina, E.; van Rooijen, W.J.; Spaan, A.N.; van Kessel, K.P.M.; Höök, M.; Rooijakkers, S.H.M. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog. 2013, 9, e1003816. [Google Scholar] [CrossRef] [PubMed]
- Thakker, M.; Park, J.S.; Carey, V.; Lee, J.C. Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect. Immun. 1998, 66, 5183–5189. [Google Scholar] [PubMed]
- Kampen, A.H.; Tollersrud, T.; Lund, A. Staphylococcus aureus capsular polysaccharide types 5 and 8 reduce killing by bovine neutrophils in vitro. Infect. Immun. 2005, 73, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Ko, Y.-P.; Liang, X.; Ross, C.L.; Liu, Q.; Murray, B.E.; Höök, M. Collagen-binding microbial surface components recognizing adhesive matrix molecule (MSCRAMM) of Gram-positive bacteria inhibit complement activation via the classical pathway. J. Biol. Chem. 2013, 288, 20520–20531. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.M.; Wu, J.; Ruyken, M.; van Domselaar, R.; Planken, K.L.; Tzekou, A.; Ricklin, D.; Lambris, J.D.; Janssen, B.J.C.; van Strijp, J.A.G.; et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat. Immunol. 2009, 10, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Haupt, K.; Reuter, M.; van den Elsen, J.; Burman, J.; Hälbich, S.; Richter, J.; Skerka, C.; Zipfel, P.F. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement Factor H and C3b. PLoS Pathog. 2008, 4, e1000250. [Google Scholar] [CrossRef] [PubMed]
- Koch, T.K.; Reuter, M.; Barthel, D.; Böhm, S.; van den Elsen, J.; Kraiczy, P.; Zipfel, P.F.; Skerka, C. Staphylococcus aureus proteins Sbi and Efb recruit human plasmin to degrade complement C3 and C3b. PLoS One 2012, 7, e47638. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.A.; Echague, C.G.; Hair, P.S.; Ward, M.D.; Nyalwidhe, J.O.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Staphylococcus aureus surface protein SdrE binds complement regulator factor H as an immune evasion tactic. PLoS One 2012, 7, e38407. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.S.; Ward, M.D.; Semmes, O.J.; Foster, T.J.; Cunnion, K.M. Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J. Infect. Dis. 2008, 198, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.S.; Echague, C.G.; Sholl, A.M.; Watkins, J.A.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 2010, 78, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Götz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Gotz, F.; Götz, F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; van Kessel, K.P.; van Strijp, J.A. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 2001, 193, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Stapels, D.A.C.; Ramyar, K.X.; Bischoff, M.; von Köckritz-Blickwede, M.; Milder, F.J.; Ruyken, M.; Eisenbeis, J.; McWhorter, W.J.; Herrmann, M.; van Kessel, K.P.M.; et al. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 13187–92. [Google Scholar] [CrossRef] [PubMed]
- Karavolos, M.H.; Horsburgh, M.J.; Ingham, E.; Foster, S.J. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology 2003, 149, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Barriere, C.; Bruckner, R.; Centeno, D.; Talon, R. Characterisation of the katA gene encoding a catalase and evidence for at least a second catalase activity in Staphylococcus xylosus, bacteria used in food fermentation. FEMS Microbiol. Lett. 2002, 216, 277–283. [Google Scholar] [CrossRef]
- Pang, Y.Y.; Schwartz, J.; Bloomberg, S.; Boyd, J.M.; Horswill, A.R.; Nauseef, W.M. Methionine sulfoxide reductases protect against oxidative stress in Staphylococcus aureus encountering exogenous oxidants and human neutrophils. J. Innate. Immun. 2014, 6, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Essex, A.; Buchanan, J.T.; Datta, V.; Hoffman, H.M.; Bastian, J.F.; Fierer, J.; Nizet, V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005, 202, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Liu, G.Y.; Yeaman, M.R.; Nast, C.C.; Proctor, R.A.; McKinnell, J.; Bayer, A.S. Carotenoid-Related Alteration of Cell Membrane Fluidity Impacts Staphylococcus aureus Susceptibility to Host Defense Peptides. Antimicrob. Agents Chemother. 2011, 55, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.R.; Libby, S.J.; Fang, F.C. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 2008, 319, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.L.; Nobre, L.S.; Vicente, J.B.; Teixeira, M.; Saraiva, L.M. Flavohemoglobin requires microaerophilic conditions for nitrosative protection of Staphylococcus aureus. FEBS Lett. 2006, 580, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.R.; Dunman, P.M.; Fang, F.C. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol 2006, 61, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Beasley, F.C.; Vinés, E.D.; Grigg, J.C.; Zheng, Q.; Liu, S.; Lajoie, G.A.; Murphy, M.E.P.; Heinrichs, D.E. Characterization of staphyloferrin A biosynthetic and transport mutants in Staphylococcus aureus. Mol. Microbiol. 2009, 72, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Beasley, F.C.; Liu, S.; Lajoie, G.A.; Heinrichs, D.E. Molecular characterization of staphyloferrin B biosynthesis in Staphylococcus aureus. Mol Microbiol 2009, 74, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Beasley, F.C.; Marolda, C.L.; Cheung, J.; Buac, S.; Heinrichs, D.E. Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by staphyloferrin A, staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect. Immun. 2011, 79, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Sebulsky, M.T.; Heinrichs, D.E. Identification and characterization of fhuD1 and fhuD2, two genes involved in iron-hydroxamate uptake in Staphylococcus aureus. J. Bacteriol. 2001, 183, 4994–5000. [Google Scholar] [CrossRef] [PubMed]
- Speziali, C.D.; Dale, S.E.; Henderson, J.A.; Vinés, E.D.; Heinrichs, D.E. Requirement of Staphylococcus aureus ATP-binding cassette-ATPase FhuC for iron-restricted growth and evidence that it functions with more than one iron transporter. J. Bacteriol. 2006, 188, 2048–2055. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Pishchany, G.; Humayun, M.; Schneewind, O.; Skaar, E.P. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol 2006, 188, 8421–8429. [Google Scholar] [CrossRef] [PubMed]
- Kehl-Fie, T.E.; Zhang, Y.; Moore, J.L.; Farrand, A.J.; Hood, M.I.; Rathi, S.; Chazin, W.J.; Caprioli, R.M.; Skaar, E.P. MntABC and MntH contribute to systemic Staphylococcus aureus infection by competing with calprotectin for nutrient manganese. Infect. Immun. 2013, 81, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of Staphylococcal alpha-Hemolysin, a Heptameric Transmembrane Pore. Science 1996, 274, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Tsompanidou, E.; Denham, E.L.; van Dijl, J.M. Phenol-soluble modulins, hellhounds from the staphylococcal virulence-factor pandemonium. Trends Microbiol. 2013, 21, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.J.; Nijland, R.; Spaan, A.N.; Kruijtzer, J.A.W.; de Haas, C.J.C.; van Strijp, J.A.G. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Wolz, C. Intersection of the stringent response and the CodY regulon in low GC Gram-positive bacteria. Int. J. Med. Microbiol. 2014, 304, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.-C.C.; Schäfer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell. Microbiol. 2014, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.S.S.; Cohen, T.S.S.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-Induced Necroptosis Is a Major Mechanism of Staphylococcus aureus Lung Damage. PLOS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef] [PubMed]
- Melehani, J.H.; James, D.B.A.; DuMont, A.L.; Torres, V.J.; Duncan, J.A. Staphylococcus aureus Leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLOS Pathog. 2015, 11, e1004970. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, D.; Gieldon, L.; Mysore, V.; Nippe, N.; Taxman, D.J.; Duncan, J.A.; Broglie, P.M.; Marketon, K.; Austermann, J.; Vogl, T.; et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J. Leukoc. Biol. 2012, 92, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Kern, J.W.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus synthesizes adenosine to escape host immune responses. J. Exp. Med. 2009, 206, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Schneewind, O.; Missiakas, D.M. Enzymatic properties of Staphylococcus aureus adenosine synthase (AdsA). BMC Biochem. 2011, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Thiel, M.; Caldwell, C.C.; Sitkovsky, M.V. The critical role of adenosine A2A receptors in downregulation of inflammation and immunity in the pathogenesis of infectious diseases. Microbes Infect. 2003, 5, 515–526. [Google Scholar] [CrossRef]
- Haskó, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Pernet, E.; Brunet, J.; Guillemot, L.; Chignard, M.; Touqui, L.; Wu, Y. Staphylococcus aureus adenosine inhibits sPLA2-IIA-mediated host killing in the airways. J. Immunol. 2015, 194, 5312–5319. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Inforzato, A.; Doni, A.; Barajon, I.; Leone, R.; Garlanda, C.; Bottazzi, B.; Mantovani, A. PTX3 as a paradigm for the interaction of pentraxins with the complement system. Semin. Immunol. 2013, 25, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Inforzato, A.; Bottazzi, B.; Garlanda, C.; Valentino, S.; Mantovani, A. Pentraxins in humoral innate immunity. Adv. Exp. Med. Biol. 2012, 946, 1–20. [Google Scholar] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Sulica, A.; Medesan, C.; Laky, M.; Onică, D.; Sjöquist, J.; Ghetie, V. Effect of protein A of Staphylococcus aureus on the binding of monomeric and polymeric IgG to Fc receptor-bearing cells. Immunology 1979, 38, 173–9. [Google Scholar] [PubMed]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Laky, M.; Sjöquist, J.; Moraru, I.; Gheţie, V. Mutual inhibition of the binding of Clq and protein A to rabbit IgG immune complexes. Mol. Immunol. 1985, 22, 1297–1302. [Google Scholar] [CrossRef]
- Patel, A.H.; Nowlan, P.; Weavers, E.D.; Foster, T. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 1987, 55, 3103–3110. [Google Scholar] [PubMed]
- Palmqvist, N.; Foster, T.; Tarkowski, A.; Josefsson, E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb. Pathog. 2002, 33, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Pauli, N.T.; Kim, H.K.; Falugi, F.; Huang, M.; Dulac, J.; Henry Dunand, C.; Zheng, N.-Y.; Kaur, K.; Andrews, S.F.; Huang, Y.; et al. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J. Exp. Med. 2014, 211, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Visai, L.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. The Sbi Protein Is a Multifunctional Immune Evasion Factor of Staphylococcus aureus. Infect. Immun. 2011, 79, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Corrigan, R.M.; van der Sluis, T.; Gründling, A.; Speziale, P.; Geoghegan, J.A.; Foster, T.J. The immune evasion protein Sbi of Staphylococcus aureusoccurs both extracellularly and anchored to the cell envelope by binding lipoteichoic acid. Mol. Microbiol. 2012, 83, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Jusko, M.; Potempa, J.; Kantyka, T.; Bielecka, E.; Miller, H.K.; Kalinska, M.; Dubin, G.; Garred, P.; Shaw, L.N.; Blom, A.M. Staphylococcal proteases aid in evasion of the human complement system. J. Innate Immun. 2014, 6, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Skerka, C. Staphylococcus aureus: the multi headed hydra resists and controls human complement response in multiple ways. Int. J. Med. Microbiol. 2014, 304, 188–94. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.L.; Ramyar, K.X.; Ricklin, D.; Lambris, J.D.; Geisbrecht, B.V. Advances in Understanding the Structure, Function, and Mechanism of the SCIN and Efb Families of Staphylococcal Immune Evasion Proteins. Adv. Exp. Med. Biol. 2012, 40, 113–133. [Google Scholar]
- Okusawa, S.; Dinarello, C.A.; Yancey, K.B.; Endres, S.; Lawley, T.J.; Frank, M.M.; Burke, J.F.; Gelfand, J.A. C5a induction of human interleukin 1. Synergistic effect with endotoxin or interferon-gamma. J. Immunol. 1987, 139, 2635–40. [Google Scholar] [PubMed]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013, 122, 3473–81. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Heim, C.E.; Angle, A.; Sanderson, S.D.; Kielian, T. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J. Immunol. 2013, 190, 2159–68. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.M.; Ruyken, M.; Roos, A.; Daha, M.R.; Presanis, J.S.; Sim, R.B.; van Wamel, W.J.B.; van Kessel, K.P.M.; van Strijp, J.A.G. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat. Immunol. 2005, 6, 920–927. [Google Scholar] [CrossRef] [PubMed]
- DuMont, A.L.; Yoong, P.; Surewaard, B.G.J.; Benson, M.A.; Nijland, R.; van Strijp, J.A.G.; Torres, V.J. Staphylococcus aureus elaborates leukocidin AB To mediate escape from within human neutrophils. Infect. Immun. 2013, 81, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Fraunholz, M.; Sinha, B. Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell. Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Lâm, T.-T.T.; Giese, B.; Chikkaballi, D.; Kühn, A.; Wolber, W.; Pané-Farré, J.; Schäfer, D.; Engelmann, S.; Fraunholz, M.; Sinha, B. Phagolysosomal integrity is generally maintained after Staphylococcus aureus invasion of nonprofessional phagocytes but is modulated by strain 6850. Infect. Immun. 2010, 78, 3392–3393. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jubrail, J.; Morris, P.; Bewley, M.A.; Stoneham, S.; Johnston, S.A.; Foster, S.J.; Peden, A.A.; Read, R.C.; Marriott, H.M.; Dockrell, D.H. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell. Microbiol. 2015. doi:http://dx.doi.org/10.1111/j.1574-6968.2002.tb11447.x. [Google Scholar] [PubMed]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; Foster, T.; Potempa, J. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 2008, 3, e1409. [Google Scholar] [CrossRef] [PubMed]
- Giese, B.; Glowinski, F.; Paprotka, K.; Dittmann, S.; Steiner, T.; Sinha, B.; Fraunholz, M.J. Expression of δ-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of β-toxin. Cell. Microbiol. 2011, 13, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Jarry, T.M.; Memmi, G.; Cheung, A.L. The expression of alpha-haemolysin is required for Staphylococcus aureus phagosomal escape after internalization in CFT-1 cells. Cell. Microbiol. 2008, 10, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Francois, P.; Liebeke, M.; Fraunholz, M.; Goerke, C.; Krismer, B.; Schrenzel, J.; Lalk, M.; Wolz, C. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 2012, 8, e1003016. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Biswas, R.; Herbert, S.; Götz, F. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect. Immun. 2006, 74, 4598–4604. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Park, B.G.; Wolf, A.J.; Brikos, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Götz, F.; Liu, G.Y.; Underhill, D.M. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 2010, 7, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Kristian, S.A.; Dürr, M.; van Strijp, J.A.G.; Neumeister, B.; Peschel, A. MprF-mediated lysinylation of phospholipids in Staphylococcus aureus leads to protection against oxygen-independent neutrophil killing. Infect. Immun. 2003, 71, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; Van Kessel, K.P.M.; van Strijp, J.A.G.; Götz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis. 2002, 186, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Rode, T.M.; Møretrø, T.; Langsrud, S.; Langsrud, O.; Vogt, G.; Holck, A. Responses of Staphylococcus aureus exposed to HCl and organic acid stress. Can. J. Microbiol. 2010, 56, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Bore, E.; Langsrud, S.; Langsrud, Ø.; Rode, T.M.; Holck, A. Acid-shock responses in Staphylococcus aureus investigated by global gene expression analysis. Microbiology 2007, 153, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Carraro-Lacroix, L.R.; Jaumouillé, V.; Fairn, G.D.; Grinstein, S. A weak base-generating system suitable for selective manipulation of lysosomal pH. Traffic 2011, 12, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Ballal, A.; Manna, A.C. Regulation of superoxide dismutase (sod) genes by SarA in Staphylococcus aureus. J. Bacteriol. 2009, 191, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, K.; Coutts, G.; Jonsson, I.-M.; Tarkowski, A.; Kokai-Kun, J.F.; Mond, J.J.; Foster, S.J. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J. Bacteriol. 2007, 189, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Mandell, G.L. Catalase, superoxide dismutase, and virulence of Staphylococcus aureus. In vitro and in vivo studies with emphasis on staphylococcal–leukocyte interaction. J. Clin. Invest. 1975, 55, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Kehl-Fie, T.E.; Chitayat, S.; Hood, M.I.; Damo, S.; Restrepo, N.; Garcia, C.; Munro, K.A.; Chazin, W.J.; Skaar, E.P. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 2011, 10, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, M.J.; Clements, M.O.; Crossley, H.; Ingham, E.; Foster, S.J. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 2001, 69, 3744–3754. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, M.J.; Ingham, E.; Foster, S.J. In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol. 2001, 183, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, M.J.; Wharton, S.J.; Cox, A.G.; Ingham, E.; Peacock, S.; Foster, S.J. MntR modulates expression of the PerR regulon and superoxide resistance in Staphylococcus aureus through control of manganese uptake. Mol. Microbiol. 2002, 44, 1269–1286. [Google Scholar] [CrossRef] [PubMed]
- VanderVen, B.C.; Yates, R.M.; Russell, D.G. Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic 2009, 10, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Kinkel, T.L.; Roux, C.M.; Dunman, P.M.; Fang, F.C. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio 2013, 4, e00696-13. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, J.R.; Heinrichs, D.E. Recent developments in understanding the iron acquisition strategies of Gram positive pathogens. FEMS Microbiol. Rev. 2015. http://dx.doi.org/10.1093/femsre/fuv009 . [Google Scholar] [PubMed]
- Hammer, N.D.; Skaar, E.P. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 2011, 65, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed]
- Sebulsky, M.T.; Hohnstein, D.; Hunter, M.D.; Heinrichs, D.E. Identification and characterization of a membrane permease involved in iron-hydroxamate transport in Staphylococcus aureus. J. Bacteriol. 2000, 182, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Pishchany, G.; Sheldon, J.R.; Dickson, C.F.; Alam, M.T.; Read, T.D.; Gell, D.A.; Heinrichs, D.E.; Skaar, E.P. IsdB-dependent hemoglobin binding is required for acquisition of heme by Staphylococcus aureus. J. Infect. Dis. 2014, 209, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Corbin, B.D.; Seeley, E.H.; Raab, A.; Feldmann, J.; Miller, M.R.; Torres, V.J.; Anderson, K.L.; Dattilo, B.M.; Dunman, P.M.; Gerads, R.; et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 2008, 319, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.; Venkatasubramanian, S.; Paidipally, P.; Barnes, P.F.; Tvinnereim, A.; Vankayalapati, R. Interleukin 22 inhibits intracellular growth of Mycobacterium tuberculosis by enhancing Calgranulin A expression. J. Infect. Dis. 2014, 209, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.; Paidipally, P.; Barnes, P.; Tvinnereim, A.; Vankayalapat, R. IL-22 enhances Calgranulin A expression by human macrophages to inhibit virulent Mycobacterium tuberculosis growth. J. Immunol. 2011, 186, 158.21. [Google Scholar]
- Hsu, K.; Chung, Y.M.; Endoh, Y.; Geczy, C.L. TLR9 ligands induce S100A8 in macrophages via a STAT3-dependent pathway which requires IL-10 and PGE2. PLoS One 2014, 9, e103629. [Google Scholar] [CrossRef] [PubMed]
- Sitthisak, S.; Knutsson, L.; Webb, J.W.; Jayaswal, R.K. Molecular characterization of the copper transport system in Staphylococcus aureus. Microbiology 2007, 153, 4274–4283. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens 2015, 4, 826-868. https://doi.org/10.3390/pathogens4040826
Flannagan RS, Heit B, Heinrichs DE. Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens. 2015; 4(4):826-868. https://doi.org/10.3390/pathogens4040826
Chicago/Turabian StyleFlannagan, Ronald S., Bryan Heit, and David E. Heinrichs. 2015. "Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus" Pathogens 4, no. 4: 826-868. https://doi.org/10.3390/pathogens4040826
APA StyleFlannagan, R. S., Heit, B., & Heinrichs, D. E. (2015). Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens, 4(4), 826-868. https://doi.org/10.3390/pathogens4040826