Evolution of Antimicrobial Peptides to Self-Assembled Peptides for Biomaterial Applications


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Research Based Strategies for the Prevention of Medical Device Related Infection
3. Current Approaches to Self-Assembling Biomaterials
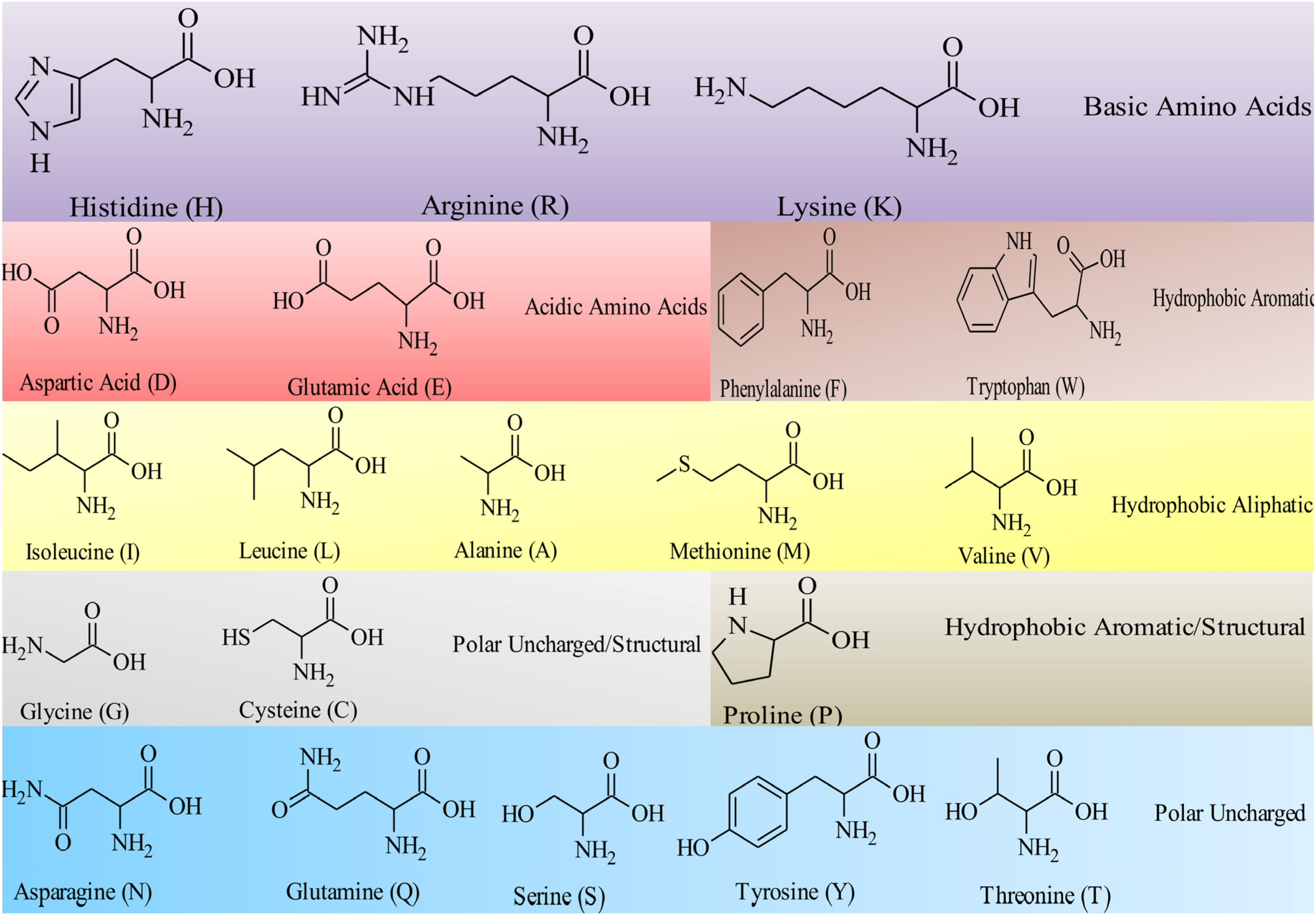

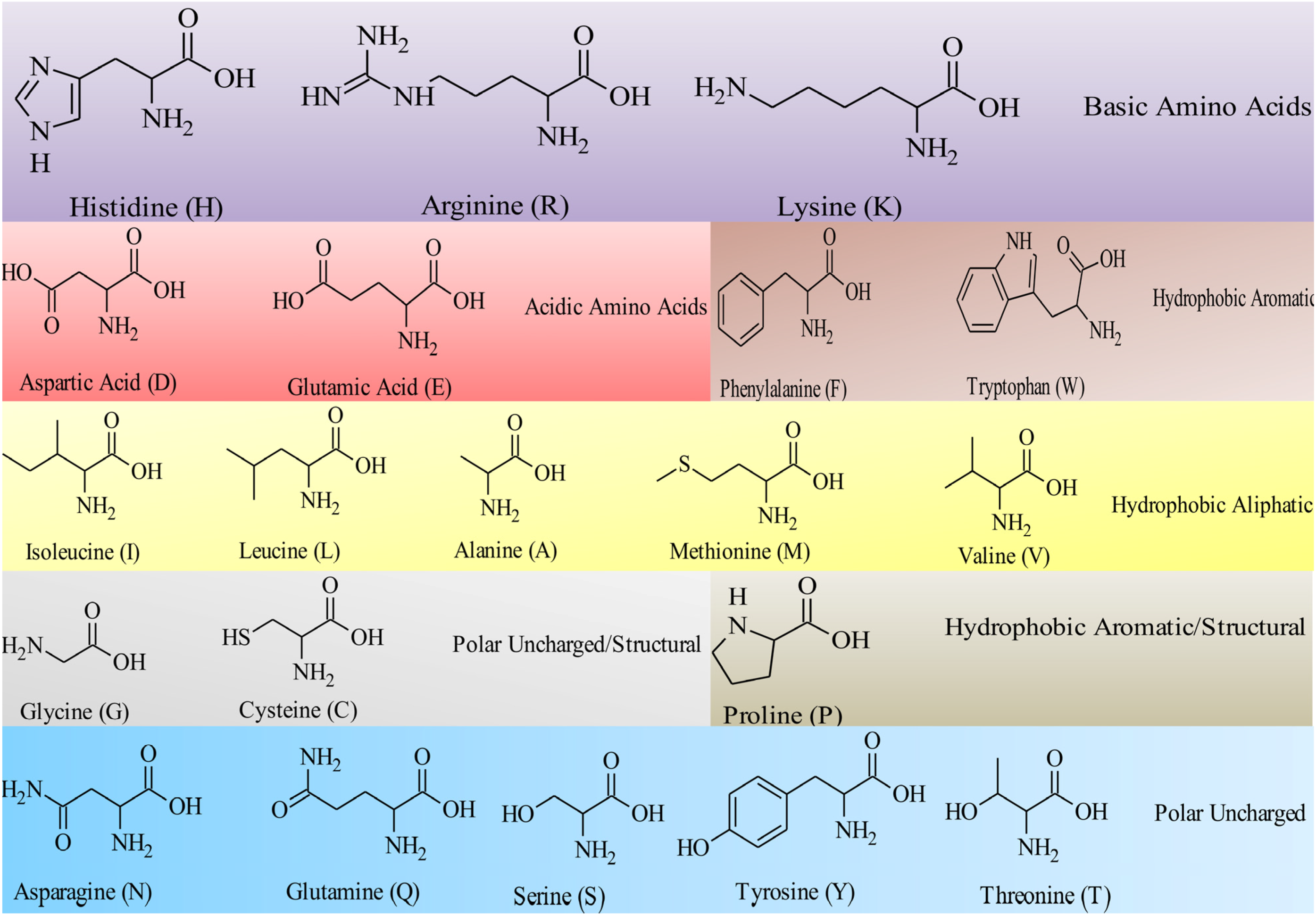
4. Antimicrobial Peptides
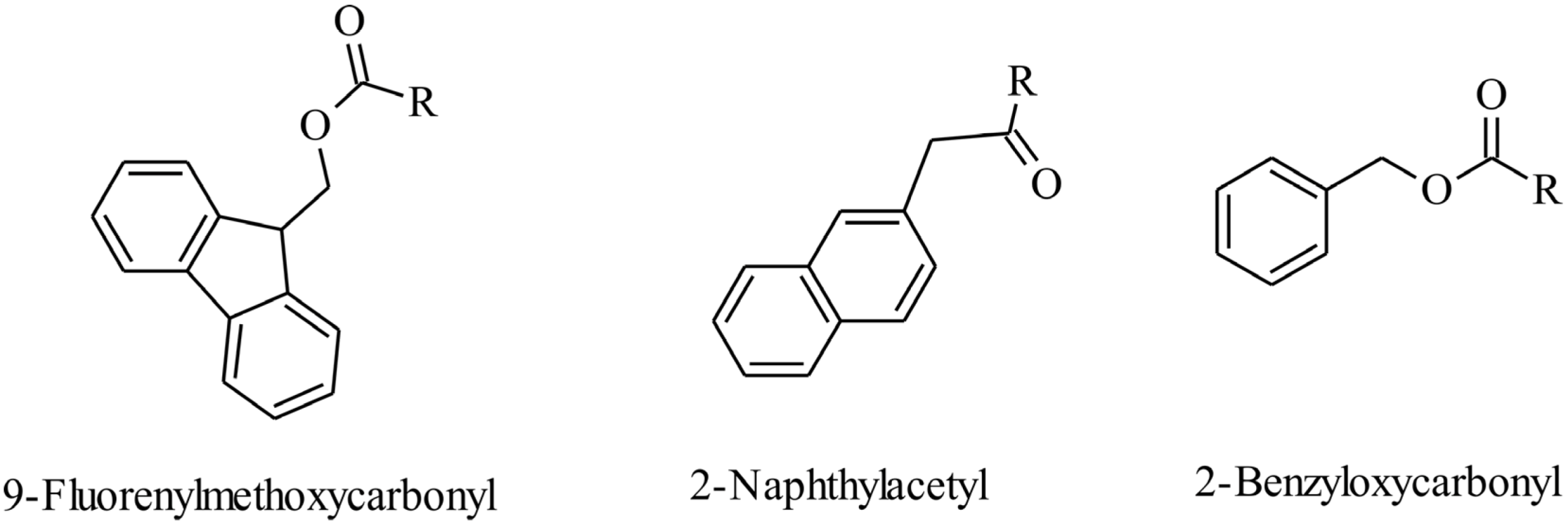
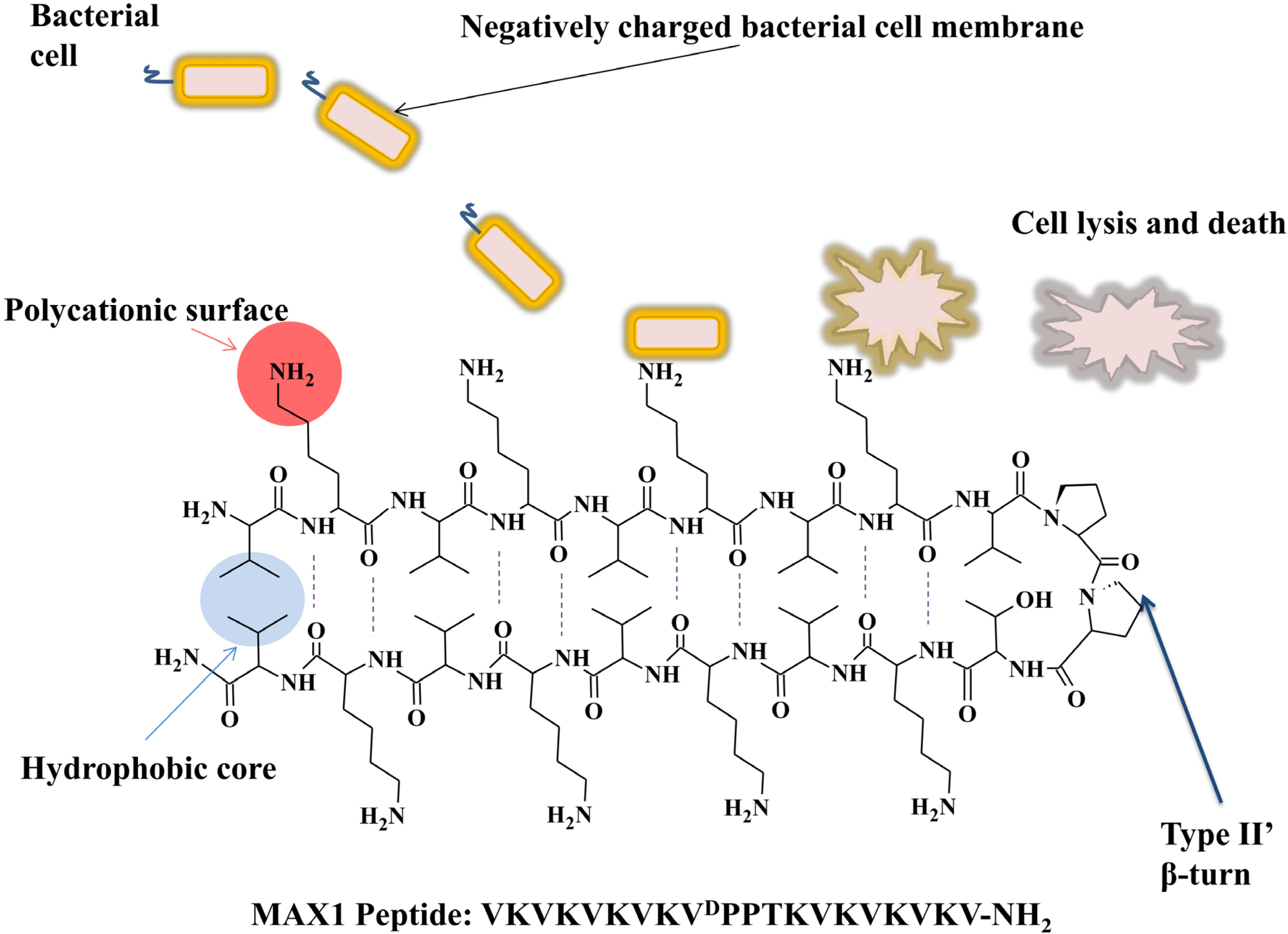
5. Self-Assembled Antimicrobial Peptides
5.1. Self-Assembly via Changes in pH and Ionic Strength
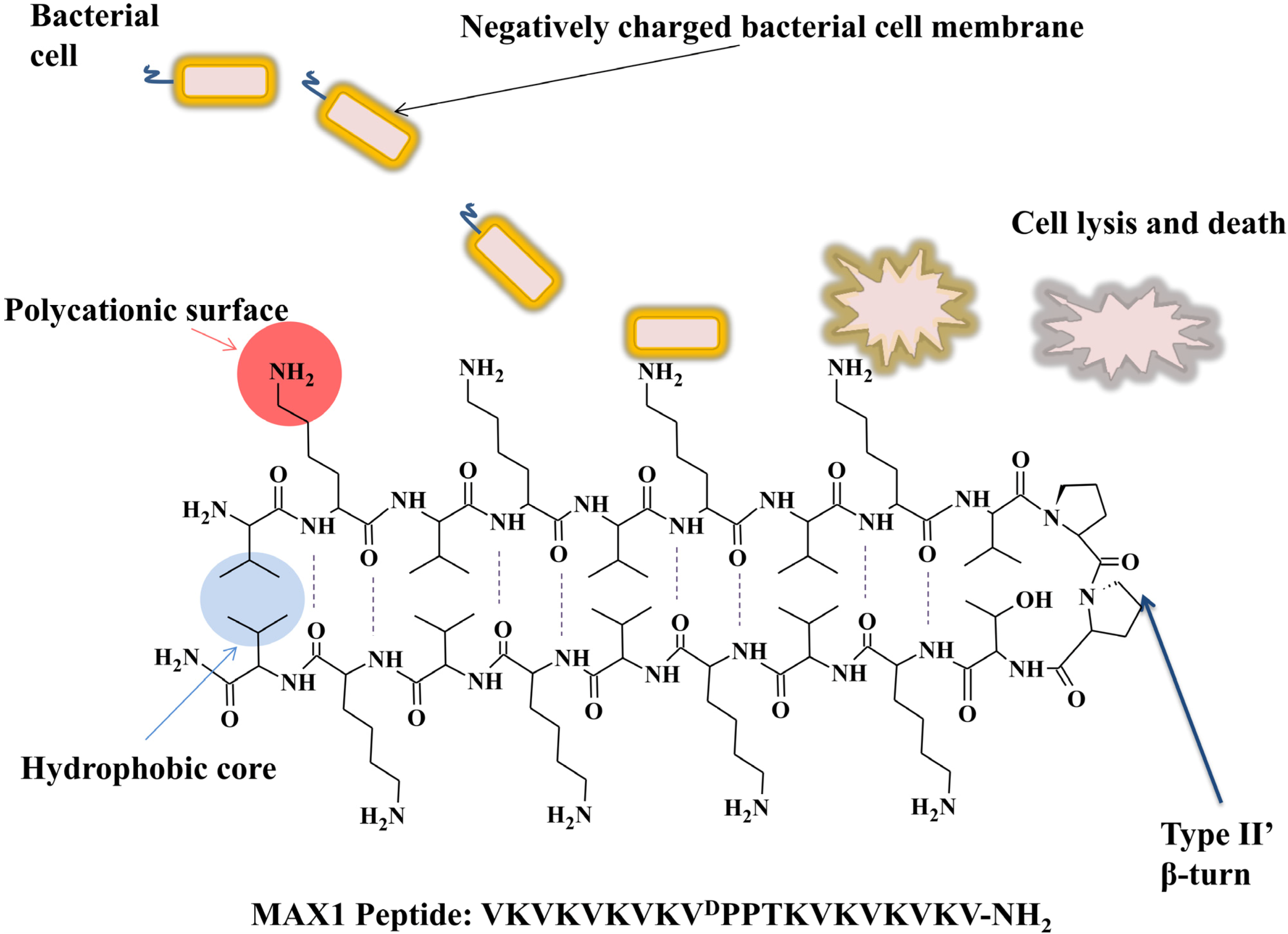
5.2. Photo-Activated Self-Assembly
5.3. Thermo-Responsive Self-Assembly
5.4. Bacterial Enzymatic Self-Assembly
6. Future Perspectivesand Translation of Peptide Self-Assembly to Antimicrobial Therapeutics
Acknowledgments
Conflicts of Interest
References
- Bryers, J.D. Medical biofilms. Biotechnol. Bioeng. 2008, 100, 1–18. [Google Scholar] [CrossRef]
- Ratner, B.D.; Hoffman, A.S.; Schoen, F.J.; Lemons, J.E. Introduction-Biomaterials Science: An Evolving, Multidisciplinary Endeavor. In Biomaterials Science; Ratner, B.D., Hoffman, A.S., Schoen, F.J., Lemons, J.E., Eds.; Academic Press Elsevier Inc.: Waltham, MA, USA, 2013; pp. 1–20. [Google Scholar]
- Kojic, E.M.; Darouiche, R.O. Candida infections of medical devices. Clin. Microbiol. Rev. 2004, 17, 255–267. [Google Scholar] [CrossRef]
- Grainger, D.W.; van der Mei, H.C.; Jutte, P.C.; van den Dungen, J.J.; Schultz, M.J.; van der Laan, B.F.; Zaat, S.A.; Busscher, H.J. Critical factors in the translation of improved antimicrobial strategies for medical implants and devices. Biomaterials 2013, 34, 9237–9243. [Google Scholar] [CrossRef]
- Zaveri, T.D.; Lewis, J.S.; Dolgova, N.V.; Clare-Salzler, M.J.; Keselowsky, B.G. Integrin-directed modulation of macrophage responses to biomaterials. Biomaterials 2014, 35, 3504–3515. [Google Scholar] [CrossRef]
- Anderson, J.M. Biological Responses to Materials. Annu. Rev. Mater. Res. 2001, 31, 81–110. [Google Scholar] [CrossRef]
- Darouiche, R.O. Treatment of infections associated with surgical implants. N. Engl. J. Med. 2004, 350, 1422–1429. [Google Scholar] [CrossRef]
- Kaplan, J.B. Antibiotic-induced biofilm formation. Int. J. Artif. Organs 2011, 34, 737–751. [Google Scholar] [CrossRef]
- Busscher, H.J.; van der Mei, H.C.; Subbiahdoss, G.; Jutte, P.C.; van den Dungen, J.J.; Zaat, S.A.; Schultz, M.J.; Grainger, D.W. Biomaterial-associated infection: Locating the finish line in the race for the surface. Sci. Transl. Med. 2012, 4, 153rv10. [Google Scholar]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef]
- Gilbert, P.; Maira-Litran, T.; McBain, A.J.; Rickard, A.H.; Whyte, F.W. The physiology and collective recalcitrance of microbial biofilm communities. Adv. Microb. Physiol. 2002, 46, 202–256. [Google Scholar]
- Francolini, I.; Donelli, G. Prevention and control of biofilm-based medical-device-related infections. FEMS Immunol. Med. Microbiol. 2010, 59, 227–238. [Google Scholar]
- Koenig, A.L.; Gambillara, V.; Grainger, D.W. Correlating fibronectin adsorption with endothelial cell adhesion and signaling on polymer substrates. J. Biomed. Mater. Res. A 2003, 64, 20–37. [Google Scholar] [CrossRef]
- MacKintosh, E.E.; Patel, J.D.; Marchant, R.E.; Anderson, J.M. Effects of biomaterial surface chemistry on the adhesion and biofilm formation of Staphylococcus epidermidis in vitro. J. Biomed. Mater. Res. A 2006, 78, 836–842. [Google Scholar] [CrossRef]
- Gottenbos, B.; van der Mei, H.C.; Busscher, H.J. Initial adhesion and surface growth of Staphylococcus epidermidis and Pseudomonas aeruginosa on biomedical polymers. J. Biomed. Mater. Res. 2000, 50, 208–214. [Google Scholar] [CrossRef]
- Hu, J.; Xu, T.; Zhu, T.; Lou, Q.; Wang, X.; Wu, Y.; Huang, R.; Liu, J.; Liu, H.; Yu, F.; et al. Monoclonal antibodies against accumulation-associated protein affect EPS biosynthesis and enhance bacterial accumulation of Staphylococcus epidermidis. PLoS One 2011, 6, e20918. [Google Scholar] [CrossRef]
- Costerton, J.W.; Montanaro, L.; Arciola, C.R. Bacterial communications in implant infections: A target for an intelligence war. Int. J. Artif. Organs 2007, 30, 757–763. [Google Scholar]
- Fux, C.A.; Costerton, J.W.; Stewart, P.S.; Stoodley, P. Survival strategies of infectious biofilms. Trends Microbiol. 2005, 13, 34–40. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilm formation: A clinically relevant microbiological process. Clin. Infect. Dis. 2001, 33, 1387–1392. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L.; Toleman, M.A.; Walsh, T.R. Does broad-spectrum beta-lactam resistance due to NDM-1 herald the end of the antibiotic era for treatment of infections caused by Gram-negative bacteria? J. Antimicrob. Chemother. 2011, 66, 689–692. [Google Scholar] [CrossRef]
- Brooks, B.D.; Brooks, A.E.; Grainger, D.W. Immunological Aspects and Antimicrobial Strategies, Antimicrobial Medical Devices in Preclinical Development and Clinical Use. In Biomaterials associated infection; Moriaty, F.T., Zaat, S.A., Busscher, H.J., Eds.; Springer: New York, NY, USA, 2013; pp. 307–354. [Google Scholar]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; WHO Press: Geneva, Switzerland, 2014. [Google Scholar]
- Crnich, C.J.; Drinka, P. Medical device-associated infections in the long-term care setting. Infect. Dis. Clin. N. Am. 2012, 26, 143–164. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilms and device-associated infections. Emerg. Infect. Dis. 2001, 7, 277–281. [Google Scholar] [CrossRef]
- Plowman, R. The socioeconomic burden of hospital acquired infection. Eur. Surveill. 2000, 5, 4. [Google Scholar]
- Pittet, D.; Allegranzi, B.; Boyce, J.; World Health Organization World Alliance for Patient Safety First Global Patient Safety Challenge Core Group of Experts. The World Health Organization Guidelines on Hand Hygiene in Health Care and their consensus recommendations. Infect. Control Hosp. Epidemiol. 2009, 30, 611–622. [Google Scholar] [CrossRef]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–492. [Google Scholar] [CrossRef]
- Saldarriaga Fernandez, I.C.; van der Mei, H.C.; Lochhead, M.J.; Grainger, D.W.; Busscher, H.J. The inhibition of the adhesion of clinically isolated bacterial strains on multi-component cross-linked poly(ethylene glycol)-based polymer coatings. Biomaterials 2007, 28, 4105–4112. [Google Scholar] [CrossRef]
- Zhang, Z.; Nix, C.A.; Ercan, U.K.; Gerstenhaber, J.A.; Joshi, S.G.; Zhong, Y. Calcium binding-mediated sustained release of minocycline from hydrophilic multilayer coatings targeting infection and inflammation. PLoS One 2014, 9, e84360. [Google Scholar] [CrossRef]
- Adams, D.J. Dipeptide and tripeptide conjugates as low-molecular-weight hydrogelators. Macromol. Biosci. 2011, 11, 160–173. [Google Scholar] [CrossRef]
- Kopecek, J.; Yang, J. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009, 5, 805–816. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Ahearn, D.G.; Grace, D.T.; Jennings, M.J.; Borazjani, R.N.; Boles, K.J.; Rose, L.J.; Simmons, R.B.; Ahanotu, E.N. Effects of hydrogel/silver coatings on in vitro adhesion to catheters of bacteria associated with urinary tract infections. Curr. Microbiol. 2000, 41, 120–125. [Google Scholar] [CrossRef]
- Wang, Y.; Burgress, D.J.; Aagaard, J. Drug Device Combination Products. In Drug Device Combination Products: Delivery Technologies and Applications; Lewis, A., Ed.; Woodhead Publishing Ltd.: Cambridge, UK, 2010; pp. 11–12. [Google Scholar]
- Gaonkar, T.A.; Modak, S.M. Comparison of microbial adherence to antiseptic and antibiotic central venous catheters using a novel agar subcutaneous infection model. J. Antimicrob. Chemother. 2003, 52, 389–396. [Google Scholar] [CrossRef]
- Yorganci, K.; Krepel, C.; Weigelt, J.A.; Edmiston, C.E. In vitro evaluation of the antibacterial activity of three different central venous catheters against Gram-positive bacteria. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 379–384. [Google Scholar]
- Gransden, W.R. Antibiotic resistance. Nosocomial Gram-negative infection. J. Med. Microbiol. 1997, 46, 436–439. [Google Scholar]
- Thomas, W.E.; Trintchina, E.; Forero, M.; Vogel, V.; Sokurenko, E.V. Bacterial adhesion to target cells enhanced by shear force. Cell 2002, 109, 913–923. [Google Scholar] [CrossRef]
- Rachid, S.; Ohlsen, K.; Witte, W.; Hacker, J.; Ziebuhr, W. Effect of subinhibitory antibiotic concentrations on polysaccharide intercellular adhesin expression in biofilm-forming Staphylococcus epidermidis. Antimicrob. Agents Chemother. 2000, 44, 3357–3363. [Google Scholar] [CrossRef]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. Biomolecular mechanisms of staphylococcal biofilm formation. Future Microbiol. 2013, 8, 509–524. [Google Scholar] [CrossRef]
- Danese, P.N. Antibiofilm approaches: Prevention of catheter colonization. Chem. Biol. 2002, 9, 873–880. [Google Scholar] [CrossRef]
- Rochford, E.T.J.; Jaekel, D.J.; Hickok, N.J.; Richards, R.G.; Moriarity, T.F.; Poulsson, A.H.C. PEEK Biomaterials Handbook; Kurtz, S.M., Ed.; Elsevier: Oxford, UK, 2012; pp. 104–105. [Google Scholar]
- Veiga, A.S.; Schneider, J.P. Antimicrobial hydrogels for the treatment of infection. Biopolymers 2013, 100, 637–644. [Google Scholar] [CrossRef]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. The potential of antimicrobial peptides as biocides. Int. J. Mol. Sci. 2011, 12, 6566–6596. [Google Scholar] [CrossRef]
- Laverty, G.; McLaughlin, M.; Shaw, C.; Gorman, S.P.; Gilmore, B.F. Antimicrobial activity of short, synthetic cationic lipopeptides. Chem. Biol. Drug Des. 2010, 75, 563–569. [Google Scholar] [CrossRef]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. Antimicrobial peptide incorporated poly(2-hydroxyethyl methacrylate) hydrogels for the prevention of Staphylococcus epidermidis-associated biomaterial infections. J. Biomed. Mater. Res. A 2012, 100, 1803–1814. [Google Scholar] [CrossRef]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals [Basel] 2013, 6, 1543–1575. [Google Scholar]
- Dasgupta, A.; Mondal, J.H.; Das, D. Peptide hydrogels. RSC Adv. 2013, 3, 9117–9149. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, G.; Guo, Z.; Guo, Z.; Xu, B. Intracellular hydrogelation of small molecules inhibits bacterial growth. Angew. Chem. Int. Ed. Engl. 2007, 46, 8216–8219. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Wang, C.; Zhao, X. Stimuli-responsive self-assembling peptides made from antibacterial peptides. Nanoscale 2013, 5, 6413–6421. [Google Scholar] [CrossRef]
- Kushner, D.J. Self-assembly of biological structures. Bacteriol. Rev. 1969, 33, 302–345. [Google Scholar]
- Zhang, S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [Google Scholar] [CrossRef]
- Stephanopoulos, N.; Ortony, J.H.; Stupp, S.I. Self-Assembly for the Synthesis of Functional Biomaterials. Acta Mater. 2013, 61, 912–930. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Javid, N.; Roy, S.; Zelzer, M.; Yang, Z.; Sefcik, J.; Ulijn, R.V. Cooperative self-assembly of peptide gelators and proteins. Biomacromolecules 2013, 14, 4368–4376. [Google Scholar] [CrossRef]
- Sargent, J.F., Jr. Nanotechnology: A policy primer. Available online: http://digitalcommons.ilr.cornell.edu/cgi/viewcontent.cgi?article=1911&context=key_workplace (accessed on 28 September 2014).
- Bohorquez, M.; Koch, C.; Trygstad, T.; Pandit, N. A Study of the Temperature-Dependent Micellization of Pluronic F127. J. Colloid Interface Sci. 1999, 216, 34–40. [Google Scholar] [CrossRef]
- Nie, S.; Hsiao, W.L.; Pan, W.; Yang, Z. Thermoreversible Pluronic F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: In vitro drug release, cell cytotoxicity, and uptake studies. Int. J. Nanomed. 2011, 6, 151–166. [Google Scholar]
- Liaw, J.; Lin, Y. Evaluation of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) (PEO-PPO-PEO) gels as a release vehicle for percutaneous fentanyl. J. Control. Release 2000, 68, 273–282. [Google Scholar] [CrossRef]
- Barichello, J.M.; Morishita, M.; Takayama, K.; Nagai, T. Absorption of insulin from pluronic F-127 gels following subcutaneous administration in rats. Int. J. Pharm. 1999, 184, 189–198. [Google Scholar] [CrossRef]
- Escobar-Chavez, J.J.; Lopez-Cervantes, M.; Naik, A.; Kalia, Y.N.; Quintanar-Guerrero, D.; Ganem-Quintanar, A. Applications of thermo-reversible Pluronic F-127 gels in pharmaceutical formulations. J. Pharm. Pharm. Sci. 2006, 9, 339–358. [Google Scholar]
- Wesenberg-Ward, K.E.; Tyler, B.J.; Sears, J.T. Adhesion and biofilm formation of Candida albicans on native and Pluronic-treated polystyrene. Biofilms 2005, 2, 63–71. [Google Scholar] [CrossRef]
- Veyries, M.L.; Faurisson, F.; Joly-Guillou, M.L.; Rouveix, B. Control of staphylococcal adhesion to polymethylmethacrylate and enhancement of susceptibility to antibiotics by poloxamer 407. Antimicrob. Agents Chemother. 2000, 44, 1093–1096. [Google Scholar] [CrossRef]
- Leszczynska, K.; Namiot, A.; Cruz, K.; Byfield, F.J.; Won, E.; Mendez, G.; Sokolowski, W.; Savage, P.B.; Bucki, R.; Janmey, P.A. Potential of ceragenin CSA-13 and its mixture with Pluronic F-127 as treatment of topical bacterial infections. J. Appl. Microbiol. 2011, 110, 229–238. [Google Scholar] [CrossRef]
- Kant, V.; Gopal, A.; Kumar, D.; Gopalkrishnan, A.; Pathak, N.N.; Kurade, N.P.; Tandan, S.K.; Kumar, D. Topical Pluronic F-127 gel application enhances cutaneous wound healing in rats. Acta Histochem. 2014, 116, 5–13. [Google Scholar] [CrossRef]
- Norris, P.; Noble, M.; Francolini, I.; Vinogradov, A.M.; Stewart, P.S.; Ratner, B.D.; Costerton, J.W.; Stoodley, P. Ultrasonically controlled release of ciprofloxacin from self-assembled coatings on poly(2-hydroxyethyl methacrylate) hydrogels for Pseudomonas aeruginosa biofilm prevention. Antimicrob. Agents Chemother. 2005, 49, 4272–4279. [Google Scholar] [CrossRef]
- Kruszewski, K.M.; Nistico, L.; Longwell, M.J.; Hynes, M.J.; Maurer, J.A.; Hall-Stoodley, L.; Gawalt, E.S. Reducing Staphylococcus aureus biofilm formation on stainless steel 316L using functionalized self-assembled monolayers. Mater. Sci. Eng. C Mater. Biol. Appl. 2013, 33, 2059–2069. [Google Scholar] [CrossRef]
- Hou, S.; Burton, E.A.; Simon, K.A.; Blodgett, D.; Luk, Y.Y.; Ren, D. Inhibition of Escherichia coli biofilm formation by self-assembled monolayers of functional alkanethiols on gold. Appl. Environ. Microbiol. 2007, 73, 4300–4307. [Google Scholar] [CrossRef]
- Wang, Q.; Uzunoglu, E.; Wu, Y.; Libera, M. Self-assembled poly(ethylene glycol)-co-acrylic acid microgels to inhibit bacterial colonization of synthetic surfaces. ACS Appl. Mater. Interfaces 2012, 4, 2498–2506. [Google Scholar] [CrossRef]
- Zhang, S.; Marini, D.M.; Hwang, W.; Santoso, S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr. Opin. Chem. Biol. 2002, 6, 865–871. [Google Scholar] [CrossRef]
- Zhang, S.; Holmes, T.; Lockshin, C.; Rich, A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA 1993, 90, 3334–3338. [Google Scholar] [CrossRef]
- Aggeli, A.; Nyrkova, I.A.; Bell, M.; Harding, R.; Carrick, L.; McLeish, T.C.; Semenov, A.N.; Boden, N. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide beta -sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. USA 2001, 98, 11857–11862. [Google Scholar] [CrossRef]
- Fields, G.B.; Tirrell, M.V. Self-Assembling Amphiphiles for Construction of Peptide Secondary Structures. Patent US 08/702,254 and WO 1998007752 A1, 2000. [Google Scholar]
- Ghadiri, M.R.; Granja, J.R.; Milligan, R.A.; McRee, D.E.; Khazanovich, N. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature 1993, 366, 324–327. [Google Scholar] [CrossRef]
- Rajagopal, K.; Schneider, J.P. Self-assembling peptides and proteins for nanotechnological applications. Curr. Opin. Struct. Biol. 2004, 14, 480–486. [Google Scholar] [CrossRef]
- Ulijn, R.V.; Smith, A.M. Designing peptide based nanomaterials. Chem. Soc. Rev. 2008, 37, 664–675. [Google Scholar] [CrossRef]
- Caplan, M.R.; Schwartzfarb, E.M.; Zhang, S.; Kamm, R.D.; Lauffenburger, D.A. Control of self-assembling oligopeptide matrix formation through systematic variation of amino acid sequence. Biomaterials 2002, 23, 219–227. [Google Scholar] [CrossRef]
- Hong, Y.; Pritzker, M.D.; Legge, R.L.; Chen, P. Effect of NaCl and peptide concentration on the self-assembly of an ionic-complementary peptide EAK16-II. Colloids Surf. B Biointerfaces 2005, 46, 152–161. [Google Scholar] [CrossRef]
- Joshi, N.S.; Whitaker, L.R.; Francis, M.B. A three-component Mannich-type reaction for selective tyrosine bioconjugation. J. Am. Chem. Soc. 2004, 126, 15942–15943. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, G.; Wang, L.; Xu, B. Using a kinase/phosphatase switch to regulate a supramolecular hydrogel and forming the supramolecular hydrogel in vivo. J. Am. Chem. Soc. 2006, 128, 3038–3043. [Google Scholar] [CrossRef]
- Mahalka, A.K.; Kinnunen, P.K. Binding of amphipathic alpha-helical antimicrobial peptides to lipid membranes: Lessons from temporins B and L. Biochim. Biophys. Acta 2009, 1788, 1600–1609. [Google Scholar] [CrossRef]
- Paramonov, S.E.; Jun, H.W.; Hartgerink, J.D. Self-assembly of peptide-amphiphile nanofibers: The roles of hydrogen bonding and amphiphilic packing. J. Am. Chem. Soc. 2006, 128, 7291–7298. [Google Scholar] [CrossRef]
- Silva, G.A.; Czeisler, C.; Niece, K.L.; Beniash, E.; Harrington, D.A.; Kessler, J.A.; Stupp, S.I. Selective differentiation of neural progenitor cells by high-epitope density nanofibers. Science 2004, 303, 1352–1355. [Google Scholar] [CrossRef]
- Gazit, E. Self-assembled peptide nanostructures: The design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 2007, 36, 1263–1269. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.I. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Burley, S.K.; Petsko, G.A. Aromatic-aromatic interaction: A mechanism of protein structure stabilization. Science 1985, 229, 23–28. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef]
- Yang, Z.; Gu, H.; Zhang, Y.; Wang, L.; Xu, B. Small molecule hydrogels based on a class of antiinflammatory agents. Chem. Commun. 2004, 2, 208–209. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, G.; Ma, M.; Gao, Y.; Xu, B. Conjugates of naphthalene and dipeptides confer molecular hydrogelators with high efficiency of hydrogelation and superhelical nanofibers. J. Mater. Chem. 2007, 17, 850–854. [Google Scholar] [CrossRef]
- Kyle, S.; Aggeli, A.; Ingham, E.; McPherson, M.J. Production of self-assembling biomaterials for tissue engineering. Trends Biotechnol. 2009, 27, 423–433. [Google Scholar] [CrossRef]
- Avrahami, D.; Shai, Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J. Biol. Chem. 2004, 279, 12277–12285. [Google Scholar] [CrossRef]
- Peschel, A.; Sahl, H.G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.; Mendoza, V.; Hancock, R.E. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 2002, 46, 605–614. [Google Scholar] [CrossRef]
- Hilpert, K.; McLeod, B.; Yu, J.; Elliott, M.R.; Rautenbach, M.; Ruden, S.; Burck, J.; Muhle-Goll, C.; Ulrich, A.S.; Keller, S.; et al. Short cationic antimicrobial peptides interact with ATP. Antimicrob. Agents Chemother. 2010, 54, 4480–4483. [Google Scholar] [CrossRef]
- Hancock, R.E.; Scott, M.G. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef]
- Finlay, B.B.; Hancock, R.E. Can innate immunity be enhanced to treat microbial infections? Nat. Rev. Microbiol. 2004, 2, 497–504. [Google Scholar]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- Grubor, B.; Meyerholz, D.K.; Ackermann, M.R. Collectins and cationic antimicrobial peptides of the respiratory epithelia. Vet. Pathol. 2006, 43, 595–612. [Google Scholar] [CrossRef]
- Brogden, K.A.; Ackermann, M.; McCray, P.B., Jr; Tack, B.F. Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob. Agents 2003, 22, 465–478. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Frick, I.M.; Andersson, E.; Tapper, H.; Bjorck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef]
- Strom, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef]
- Bisht, G.S.; Rawat, D.S.; Kumar, A.; Kumar, R.; Pasha, S. Antimicrobial activity of rationally designed amino terminal modified peptides. Bioorg. Med. Chem. Lett. 2007, 17, 4343–4346. [Google Scholar] [CrossRef]
- Makovitzki, A.; Avrahami, D.; Shai, Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef]
- Chen, Y.; Vasil, A.I.; Rehaume, L.; Mant, C.T.; Burns, J.L.; Vasil, M.L.; Hancock, R.E.; Hodges, R.S. Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem. Biol. Drug Des. 2006, 67, 162–173. [Google Scholar] [CrossRef]
- Straus, S.K.; Hancock, R.E. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: Comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 2006, 1758, 1215–1223. [Google Scholar] [CrossRef]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef]
- Levine, D.P.; Lamp, K.C. Daptomycin in the treatment of patients with infective endocarditis: Experience from a registry. Am. J. Med. 2007, 120, S28–S33. [Google Scholar] [CrossRef]
- Li, J.; Nation, R.L.; Turnidge, J.D.; Milne, R.W.; Coulthard, K.; Rayner, C.R.; Paterson, D.L. Colistin: The re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 2006, 6, 589–601. [Google Scholar] [CrossRef]
- Di Luca, M.; Maccari, G.; Nifosi, R. Treatment of microbial biofilms in the post-antibiotic era: Prophylactic and therapeutic use of antimicrobial peptides and their design by bioinformatics tools. Pathog. Dis. 2014, 70, 257–270. [Google Scholar] [CrossRef]
- Park, S.C.; Park, Y.; Hahm, K.S. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int. J. Mol. Sci. 2011, 12, 5971–5992. [Google Scholar] [CrossRef]
- Cirioni, O.; Silvestri, C.; Ghiselli, R.; Orlando, F.; Riva, A.; Mocchegiani, F.; Chiodi, L.; Castelletti, S.; Gabrielli, E.; Saba, V.; et al. Protective effects of the combination of alpha-helical antimicrobial peptides and rifampicin in three rat models of Pseudomonas aeruginosa infection. J. Antimicrob. Chemother. 2008, 62, 1332–1338. [Google Scholar] [CrossRef]
- Milne, K.E.; Gould, I.M. Combination antimicrobial susceptibility testing of multidrug-resistant Stenotrophomonas maltophilia from cystic fibrosis patients. Antimicrob. Agents Chemother. 2012, 56, 4071–4077. [Google Scholar] [CrossRef]
- Medina-Polo, J.; Jimenez-Alcaide, E.; Garcia-Gonzalez, L.; Guerrero-Ramos, F.; Perez-Cadavid, S.; Arrebola-Pajares, A.; Sopena-Sutil, R.; Benitez-Salas, R.; Diaz-Gonzalez, R.; Tejido-Sanchez, A. Healthcare-associated infections in a department of urology: Incidence and patterns of antibiotic resistance. Scand. J. Urol. 2014, 48, 203–209. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Haug, B.E.; Stensen, W.; Stiberg, T.; Svendsen, J.S. Bulky nonproteinogenic amino acids permit the design of very small and effective cationic antibacterial peptides. J. Med. Chem. 2004, 47, 4159–4162. [Google Scholar] [CrossRef]
- Pasupuleti, M.; Schmidtchen, A.; Chalupka, A.; Ringstad, L.; Malmsten, M. End-tagging of ultra-short antimicrobial peptides by W/F stretches to facilitate bacterial killing. PLoS One 2009, 4, e5285. [Google Scholar] [CrossRef]
- Rotem, S.; Radzishevsky, I.S.; Bourdetsky, D.; Navon-Venezia, S.; Carmeli, Y.; Mor, A. Analogous oligo-acyl-lysines with distinct antibacterial mechanisms. FASEB J. 2008, 22, 2652–2661. [Google Scholar] [CrossRef]
- Rotem, S.; Raz, N.; Kashi, Y.; Mor, A. Bacterial capture by peptide-mimetic oligoacyllysine surfaces. Appl. Environ. Microbiol. 2010, 76, 3301–3307. [Google Scholar] [CrossRef]
- Livne, L.; Epand, R.F.; Papahadjopoulos-Sternberg, B.; Epand, R.M.; Mor, A. OAK-based cochleates as a novel approach to overcome multidrug resistance in bacteria. FASEB J. 2010, 24, 5092–5101. [Google Scholar] [CrossRef]
- Sarig, H.; Ohana, D.; Epand, R.F.; Mor, A.; Epand, R.M. Functional studies of cochleate assemblies of an oligo-acyl-lysyl with lipid mixtures for combating bacterial multidrug resistance. FASEB J. 2011, 25, 3336–3343. [Google Scholar] [CrossRef]
- Jain, A.; Jain, A.; Gulbake, A.; Shilpi, S.; Hurkat, P.; Jain, S.K. Peptide and protein delivery using new drug delivery systems. Crit. Rev. Ther. Drug Carrier Syst. 2013, 30, 293–329. [Google Scholar] [CrossRef]
- Karin, N.; Binah, O.; Grabie, N.; Mitchell, D.J.; Felzen, B.; Solomon, M.D.; Conlon, P.; Gaur, A.; Ling, N.; Steinman, L. Short peptide-based tolerogens without self-antigenic or pathogenic activity reverse autoimmune disease. J. Immunol. 1998, 160, 5188–5194. [Google Scholar]
- Mack, D.; Rohde, H.; Harris, L.G.; Davies, A.P.; Horstkotte, M.A.; Knobloch, J.K. Biofilm formation in medical device-related infection. Int. J. Artif. Organs 2006, 29, 343–359. [Google Scholar]
- Rughani, R.V.; Salick, D.A.; Lamm, M.S.; Yucel, T.; Pochan, D.J.; Schneider, J.P. Folding, self-assembly, and bulk material properties of a de novo designed three-stranded beta-sheet hydrogel. Biomacromolecules 2009, 10, 1295–1304. [Google Scholar] [CrossRef]
- Haines, L.A.; Rajagopal, K.; Ozbas, B.; Salick, D.A.; Pochan, D.J.; Schneider, J.P. Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2005, 127, 17025–17029. [Google Scholar] [CrossRef]
- Hughes, M.; Debnath, S.; Knapp, C.W.; Ulijn, R.V. Antimicrobial properties of enzymatically triggered self-assembling aromatic peptide amphiphiles. Biomater. Sci. 2013, 1, 1138–1142. [Google Scholar] [CrossRef]
- Schneider, J.P.; Pochan, D.J.; Ozbas, B.; Rajagopal, K.; Pakstis, L.; Kretsinger, J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [Google Scholar] [CrossRef]
- Kretsinger, J.K.; Haines, L.A.; Ozbas, B.; Pochan, D.J.; Schneider, J.P. Cytocompatibility of self-assembled beta-hairpin peptide hydrogel surfaces. Biomaterials 2005, 26, 5177–5186. [Google Scholar] [CrossRef]
- Haines-Butterick, L.; Rajagopal, K.; Branco, M.; Salick, D.; Rughani, R.; Pilarz, M.; Lamm, M.S.; Pochan, D.J.; Schneider, J.P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. USA 2007, 104, 7791–7796. [Google Scholar] [CrossRef]
- Veiga, A.S.; Sinthuvanich, C.; Gaspar, D.; Franquelim, H.G.; Castanho, M.A.; Schneider, J.P. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials 2012, 33, 8907–8916. [Google Scholar] [CrossRef]
- Yokoi, H.; Kinoshita, T.; Zhang, S. Dynamic reassembly of peptide RADA16 nanofiber scaffold. Proc. Natl. Acad. Sci. USA 2005, 102, 8414–8419. [Google Scholar] [CrossRef]
- Schneider, A.; Garlick, J.A.; Egles, C. Self-assembling peptide nanofiber scaffolds accelerate wound healing. PLoS One 2008, 3, e1410. [Google Scholar] [CrossRef]
- Debnath, S.; Shome, A.; Das, D.; Das, P.K. Hydrogelation through self-assembly of fmoc-peptide functionalized cationic amphiphiles: Potent antibacterial agent. J. Phys. Chem. B 2010, 114, 4407–4415. [Google Scholar] [CrossRef]
- Williams, G.J.; Stickler, D.J. Some observations on the migration of Proteus mirabilis and other urinary tract pathogens over foley catheters. Infect. Control Hosp. Epidemiol. 2008, 29, 443–445. [Google Scholar] [CrossRef]
- McCoy, C.P.; Craig, R.A.; McGlinchey, S.M.; Carson, L.; Jones, D.S.; Gorman, S.P. Surface localisation of photosensitisers on intraocular lens biomaterials for prevention of infectious endophthalmitis and retinal protection. Biomaterials 2012, 33, 7952–7958. [Google Scholar] [CrossRef]
- Pochan, D.J.; Schneider, J.P.; Kretsinger, J.; Ozbas, B.; Rajagopal, K.; Haines, L. Thermally reversible hydrogels via intramolecular folding and consequent self-assembly of a de novo designed peptide. J. Am. Chem. Soc. 2003, 125, 11802–11803. [Google Scholar] [CrossRef]
- Tang, C.; Ulijn, R.V.; Saiani, A. Effect of glycine substitution on Fmoc-diphenylalanine self-assembly and gelation properties. Langmuir 2011, 27, 14438–14449. [Google Scholar] [CrossRef]
- Tang, C.; Ulijn, R.V.; Saiani, A. Self-assembly and gelation properties of glycine/leucine Fmoc-dipeptides. Eur. Phys. J. E. 2013, 36, 111. [Google Scholar]
- Wu, L.Q.; Payne, G.F. Biofabrication: Using biological materials and biocatalysts to construct nanostructured assemblies. Trends Biotechnol. 2004, 22, 593–599. [Google Scholar] [CrossRef]
- Hughes, M.; Frederix, P.W.J.M.; Raeburn, J.; Birchall, L.S.; Sadownik, J.; Coomer, F.C.; Lin, I.H.; Cussen, E.J.; Hunt, N.T.; Tuttle, T.; et al. Sequence/structure relationships in aromatic dipeptide hydrogels formed under thermodynamic control by enzyme-assisted self-assembly. Soft Matter 2012, 8, 5595–5602. [Google Scholar] [CrossRef]
- Hughes, M.; Birchall, L.S.; Zuberi, K.; Aitken, L.A.; Debnath, S.; Javida, N.; Ulijn, R.V. Differential supramolecular organization of Fmoc-dipeptides with hydrophilic terminal amino acid residues by biocatalytic self-assembly. Soft Matter 2012, 8, 11565–11574. [Google Scholar] [CrossRef]
- Toledano, S.; Williams, R.J.; Jayawarna, V.; Ulijn, R.V. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc. 2006, 128, 1070–1071. [Google Scholar] [CrossRef]
- Wang, H.; Yang, C.; Tan, M.; Wang, L.; Konga, D.; Yang, Z. A structure–gelation ability study in a short peptide-based ‘Super Hydrogelator’ system. Soft Matter 2011, 7, 1070–1071. [Google Scholar]
- Derman, A.I.; Beckwith, J. Escherichia coli alkaline phosphatase localized to the cytoplasm slowly acquires enzymatic activity in cells whose growth has been suspended: A caution for gene fusion studies. J. Bacteriol. 1995, 177, 3764–3770. [Google Scholar]
- Yang, K.; Metcalf, W.W. A new activity for an old enzyme: Escherichia coli bacterial alkaline phosphatase is a phosphite-dependent hydrogenase. Proc. Natl. Acad. Sci. USA 2004, 101, 7919–7924. [Google Scholar] [CrossRef]
- Yang, Z.; Ho, P.K.; Liang, G.; Chow, K.H.; Wang, Q.; Cao, Y.; Guo, Z.; Xu, B. Using β-lactamase to trigger supramolecular hydrogelation. J. Am. Chem. Soc. 2007, 129, 266–267. [Google Scholar] [CrossRef]
- Valery, C.; Paternostre, M.; Robert, B.; Gulik-Krzywicki, T.; Narayanan, T.; Dedieu, J.C.; Keller, G.; Torres, M.L.; Cherif-Cheikh, R.; Calvo, P.; et al. Biomimetic organization: Octapeptide self-assembly into nanotubes of viral capsid-like dimension. Proc. Natl. Acad. Sci. USA 2003, 100, 10258–10262. [Google Scholar] [CrossRef]
- Marchesan, S.; Qu, Y.; Waddington, L.J.; Easton, C.D.; Glattauer, V.; Lithgow, T.J.; McLean, K.M.; Forsythe, J.S.; Hartley, P.G. Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel. Biomaterials 2013, 34, 3678–3687. [Google Scholar] [CrossRef]
- Paladini, F.; Meikle, S.T.; Cooper, I.R.; Lacey, J.; Perugini, V.; Santin, M. Silver-doped self-assembling di-phenylalanine hydrogels as wound dressing biomaterials. J. Mater. Sci. Mater. Med. 2013, 24, 2461–2472. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, L.; Guan, S.; Shi, G.; Luo, Q.; Miao, L.; Thistlethwaite, I.; Huang, Z.; Xu, J.; Liu, J. Silver mineralization on self-assembled peptide nanofibers for long term antimicrobial effect. J. Mater. Chem. 2012, 22, 2575–2581. [Google Scholar] [CrossRef]
- Liu, L.; Xu, K.; Wang, H.; Tan, P.K.; Fan, W.; Venkatraman, S.S.; Li, L.; Yang, Y.Y. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat. Nanotechnol. 2009, 4, 457–463. [Google Scholar] [CrossRef]
- Laverty, G.; McCloskey, A.; Gilmore, B.F.; Jones, D.S.; Zhou, J.; Xu, B. Ultrashort cationic naphthalene-derived self-Assembled peptides as antimicrobial nanomaterials. Biomacromolecules 2014, 15, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCloskey, A.P.; Gilmore, B.F.; Laverty, G. Evolution of Antimicrobial Peptides to Self-Assembled Peptides for Biomaterial Applications. Pathogens 2014, 3, 791-821. https://doi.org/10.3390/pathogens3040791
McCloskey AP, Gilmore BF, Laverty G. Evolution of Antimicrobial Peptides to Self-Assembled Peptides for Biomaterial Applications. Pathogens. 2014; 3(4):791-821. https://doi.org/10.3390/pathogens3040791
Chicago/Turabian StyleMcCloskey, Alice P., Brendan F. Gilmore, and Garry Laverty. 2014. "Evolution of Antimicrobial Peptides to Self-Assembled Peptides for Biomaterial Applications" Pathogens 3, no. 4: 791-821. https://doi.org/10.3390/pathogens3040791
APA StyleMcCloskey, A. P., Gilmore, B. F., & Laverty, G. (2014). Evolution of Antimicrobial Peptides to Self-Assembled Peptides for Biomaterial Applications. Pathogens, 3(4), 791-821. https://doi.org/10.3390/pathogens3040791