The Natural Antimicrobial Enzyme Lysozyme is Up-Regulated in Gastrointestinal Inflammatory Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
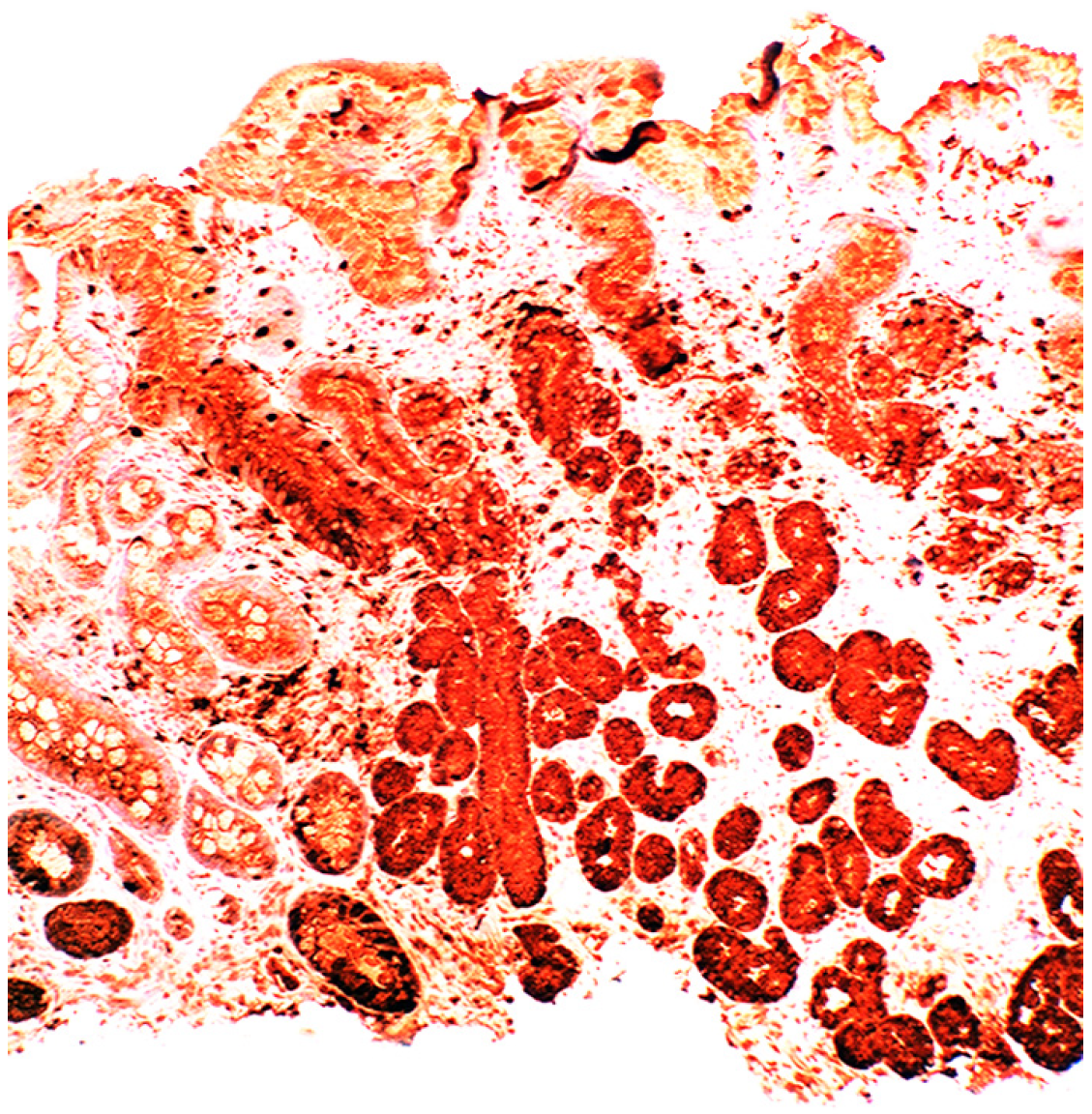{kind=link}
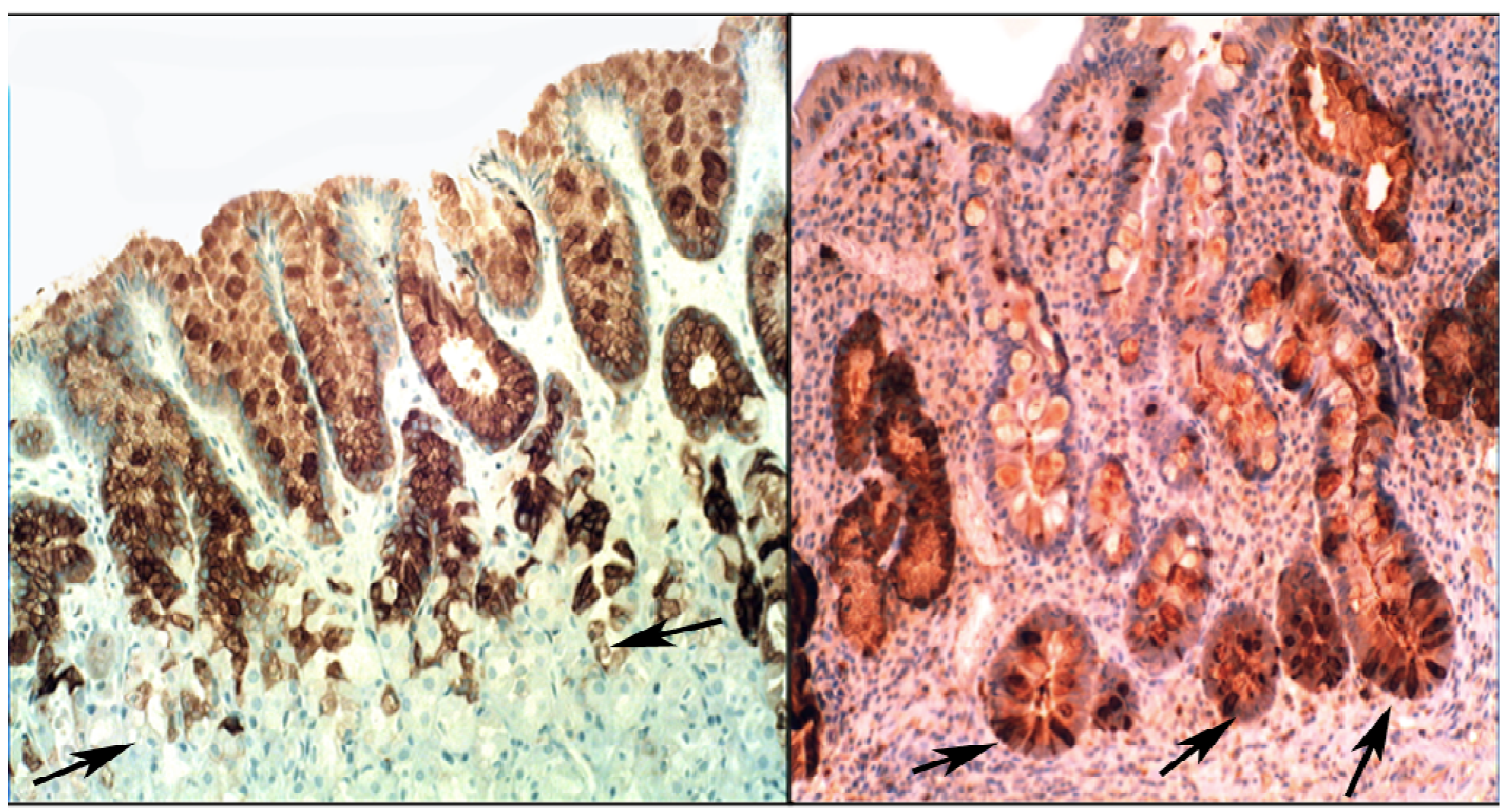{kind=link}
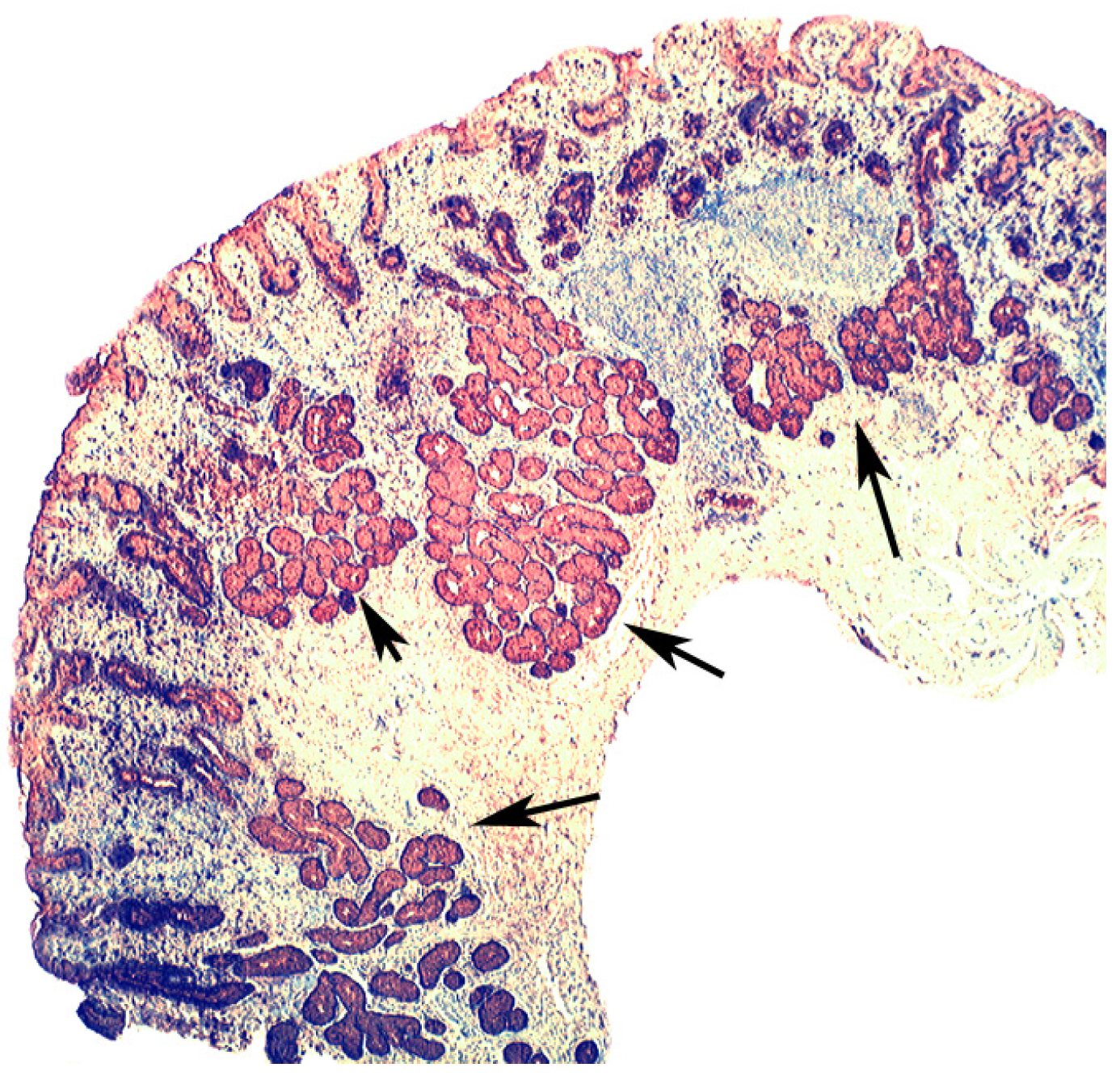{kind=link}
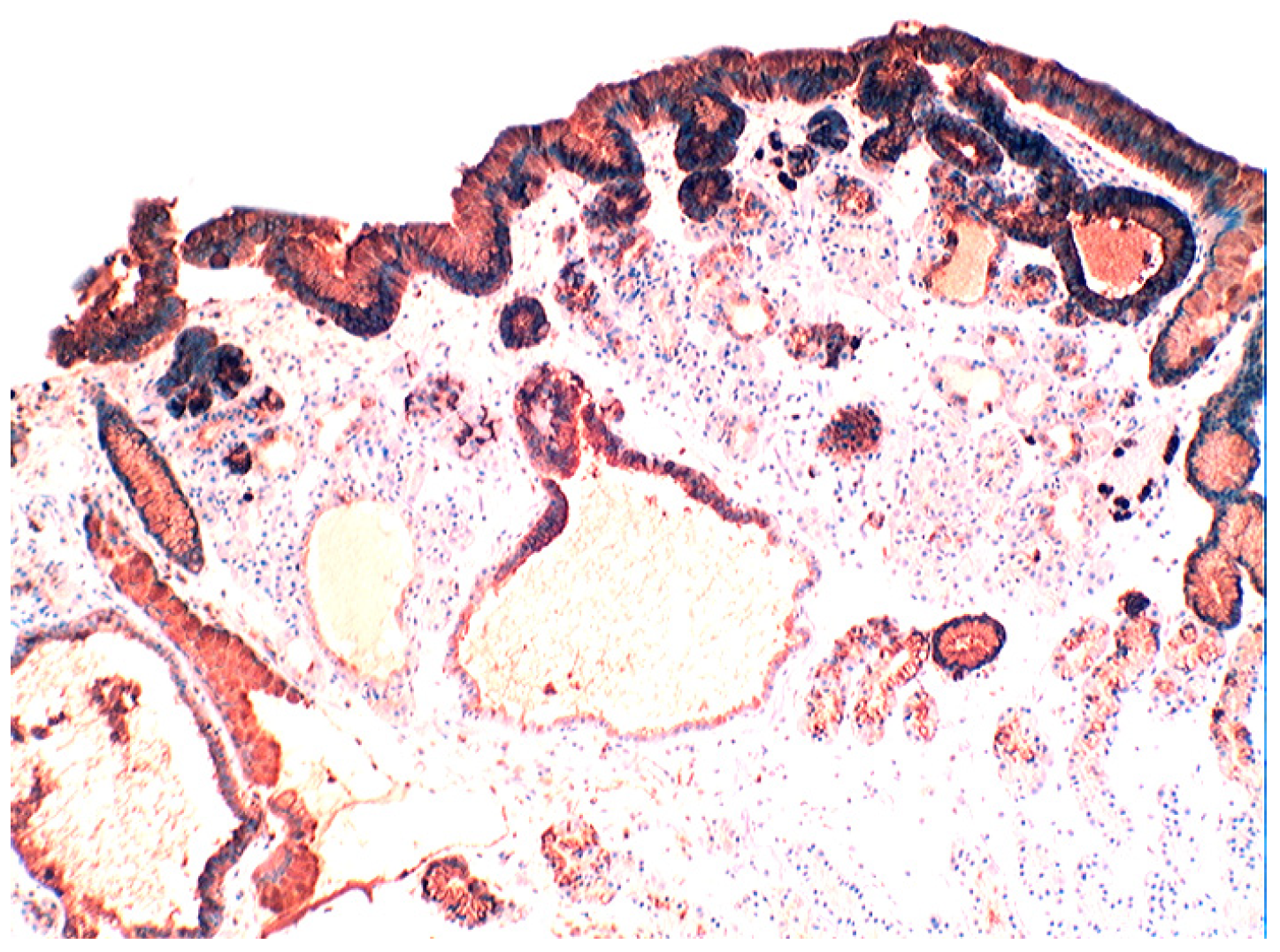{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Estimated Bacterial Flora in the GI Tract
1.2. The Discovery of Lysozyme
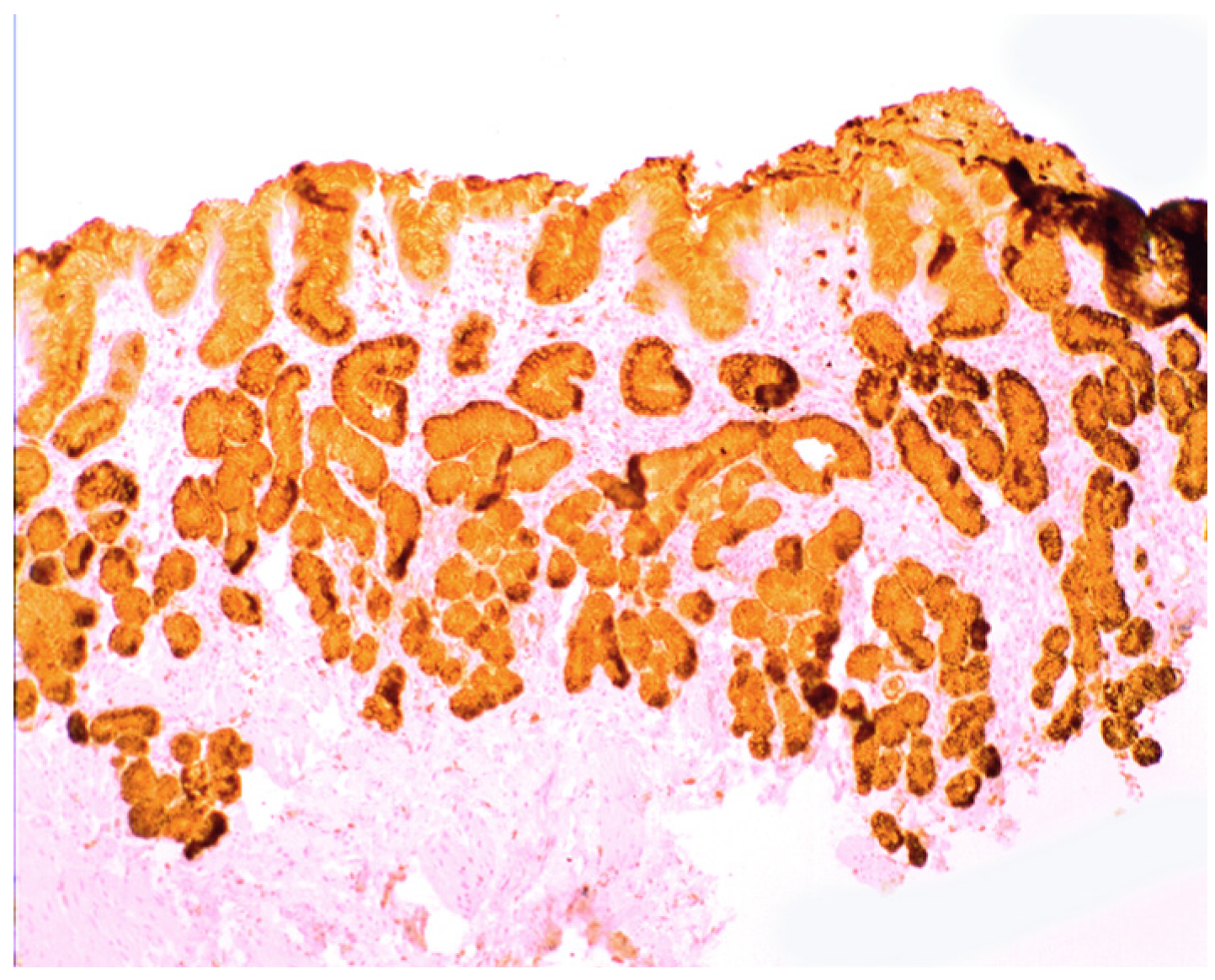
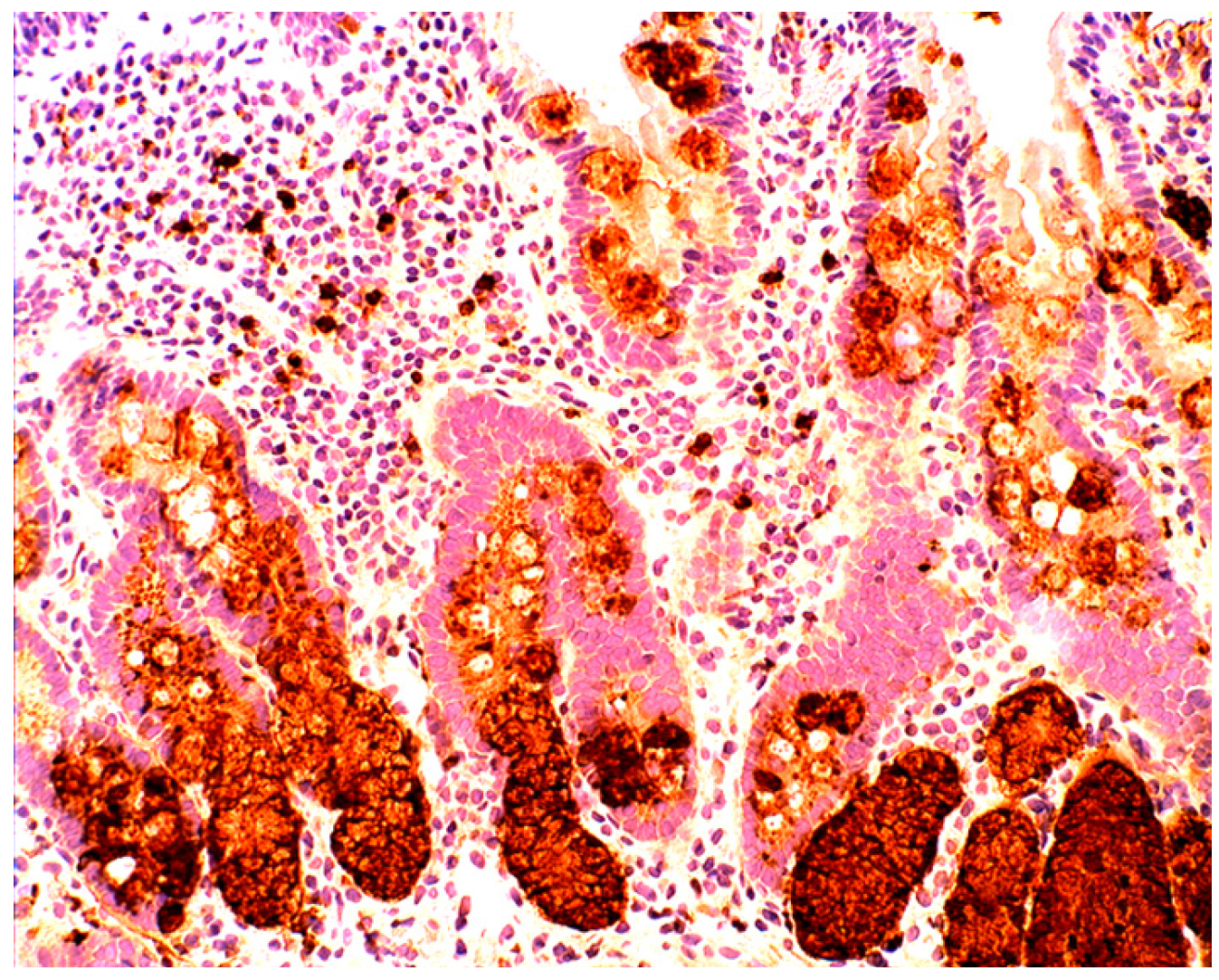
1.3. Lysozyme Is Up-Regulated in the GI Tract with Inflammation
2. Barrett’s Oesophagus
2.1. Bacteria in Barrett’s Oesophagus
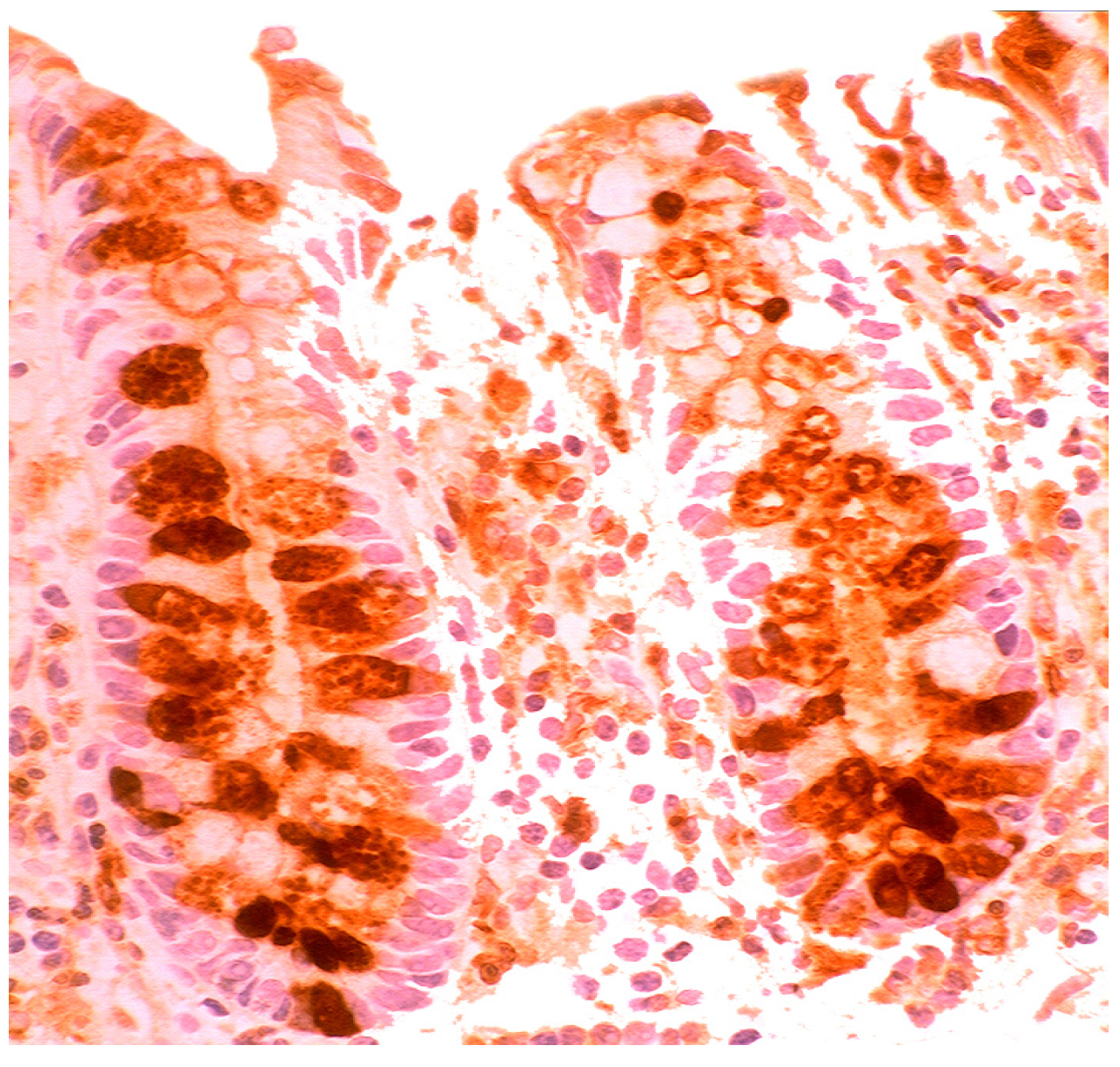
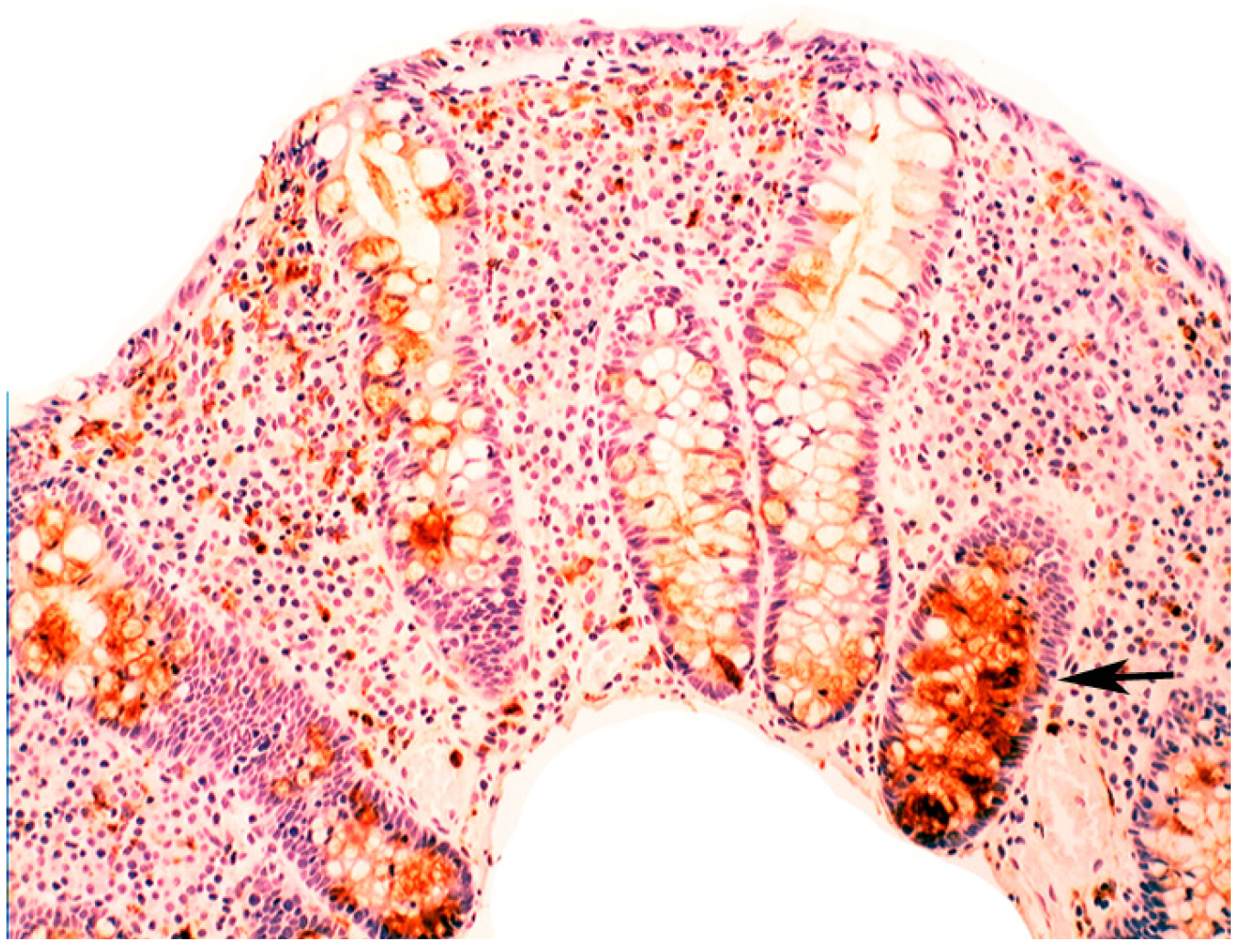
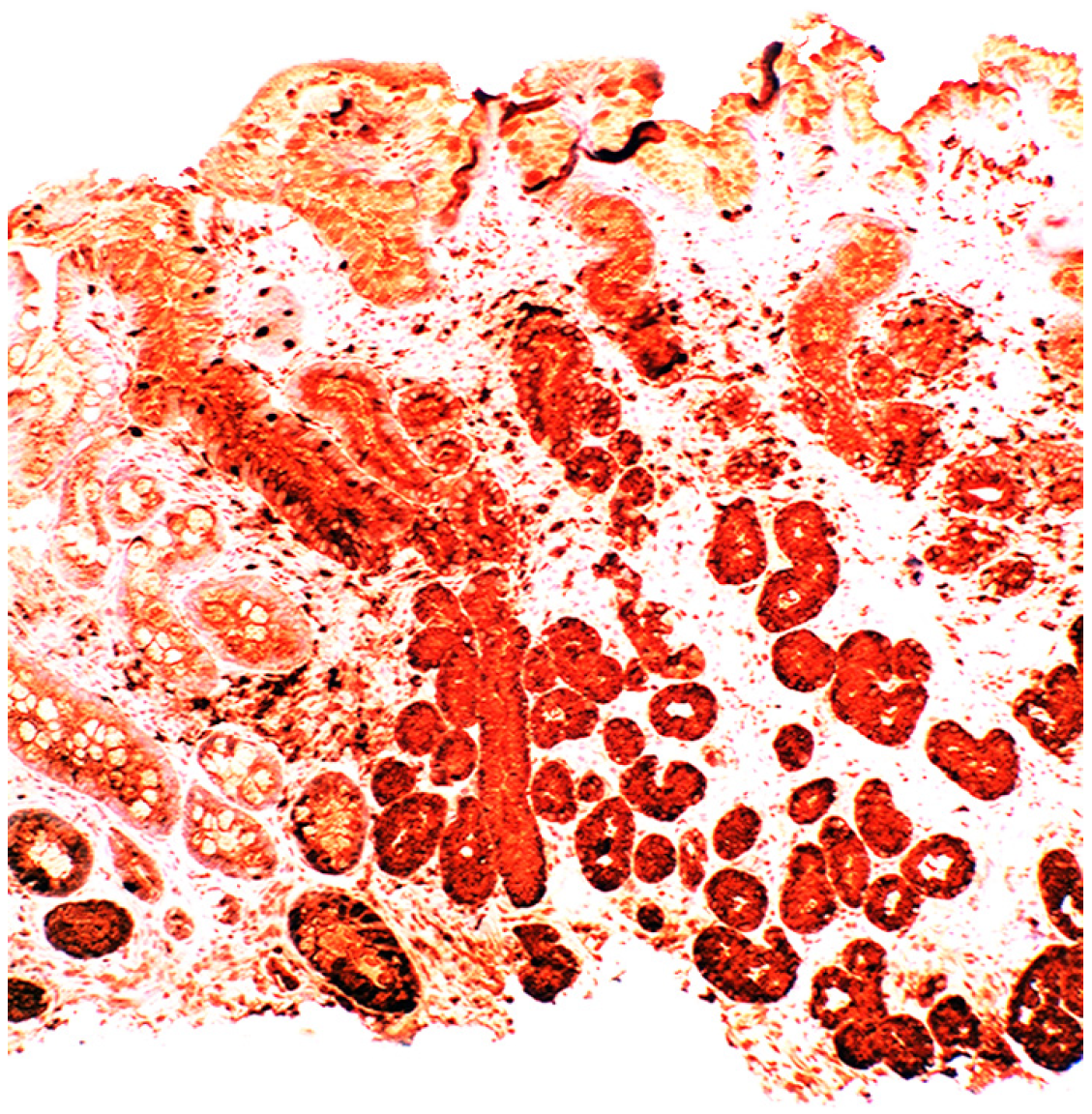
2.2. Lysozyme Is Up-Regulated in Barrett’s Oesophagitis
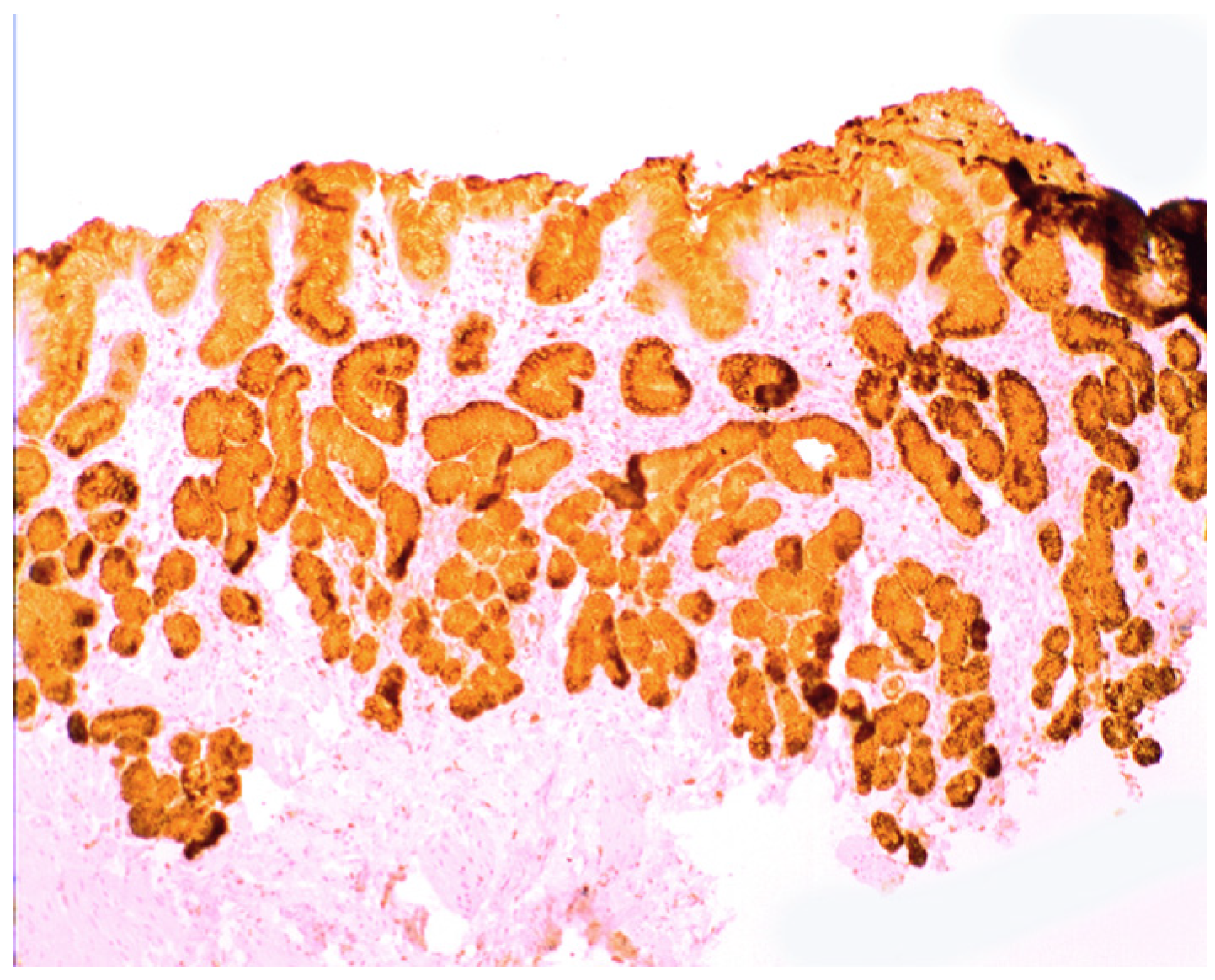

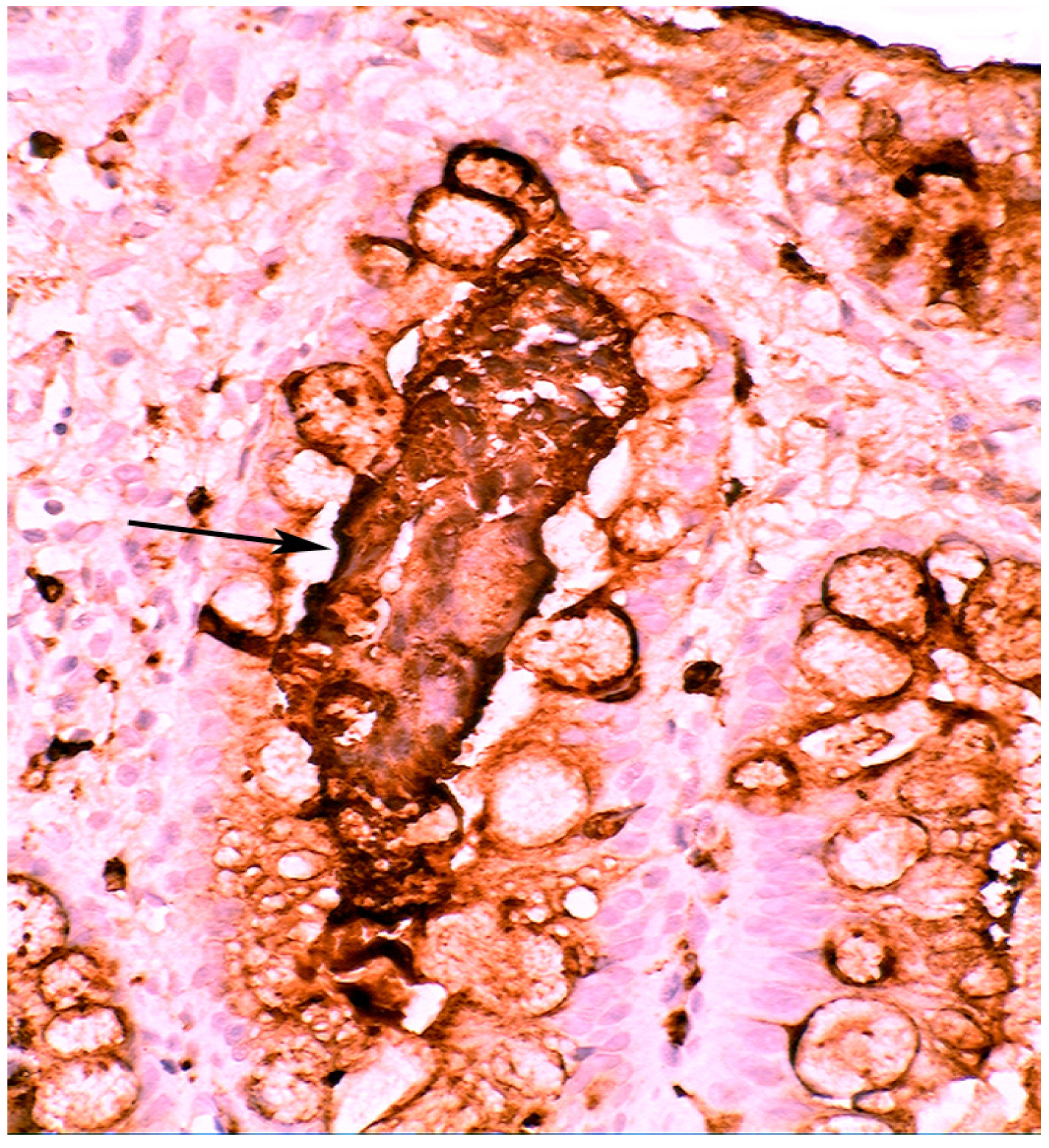
3. Chronic Gastritis
3.1. Antral Predominant Chronic Gastritis
3.2. Bacteria in Chronic Gastritis
3.3. Is Chronic Atrophic Gastritis Specifically Evoked by H. pylori and/or by Other Mucosal Insults?
3.4. Bacteria in Corpus Predominant, Autoimmune, Chronic Gastritis
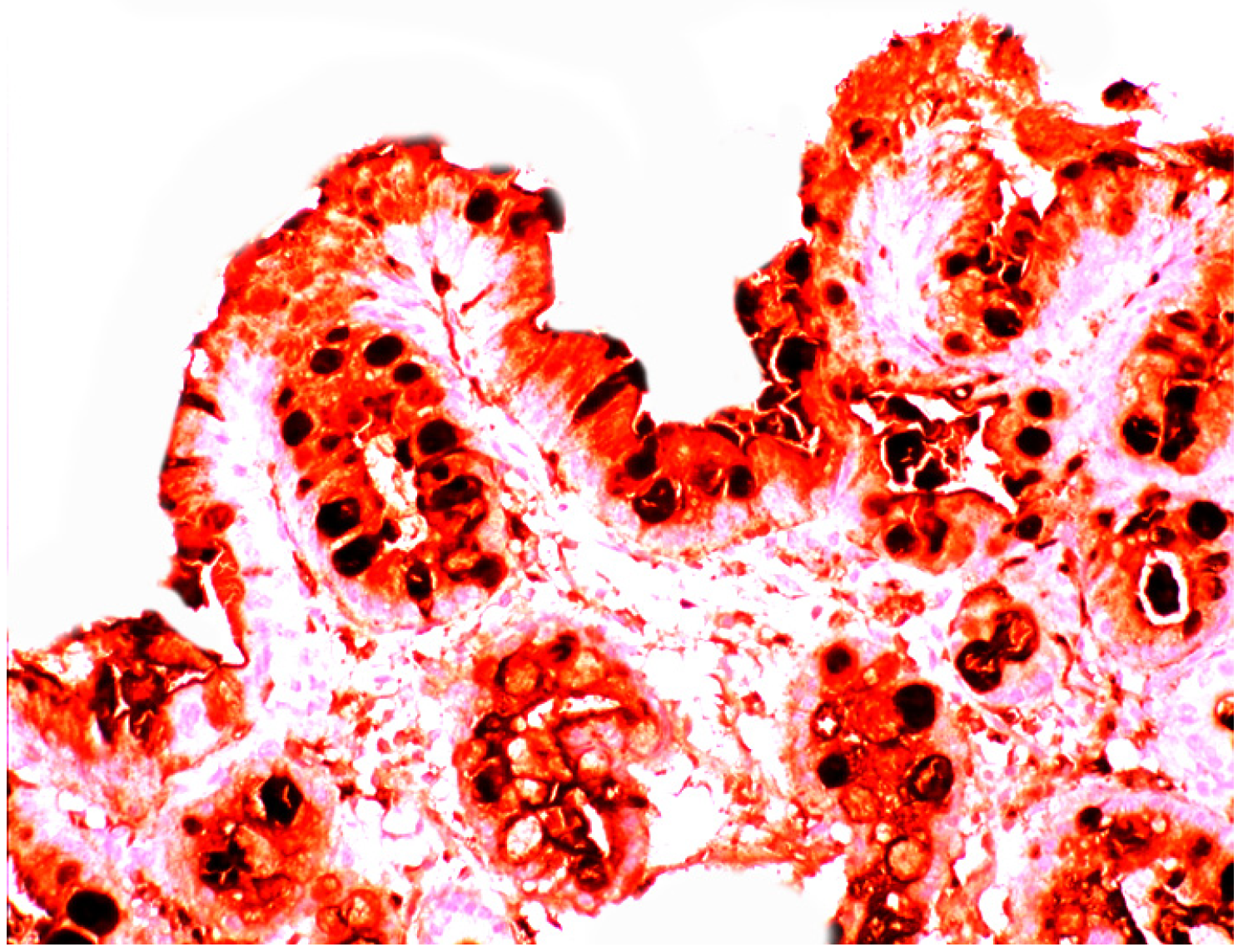
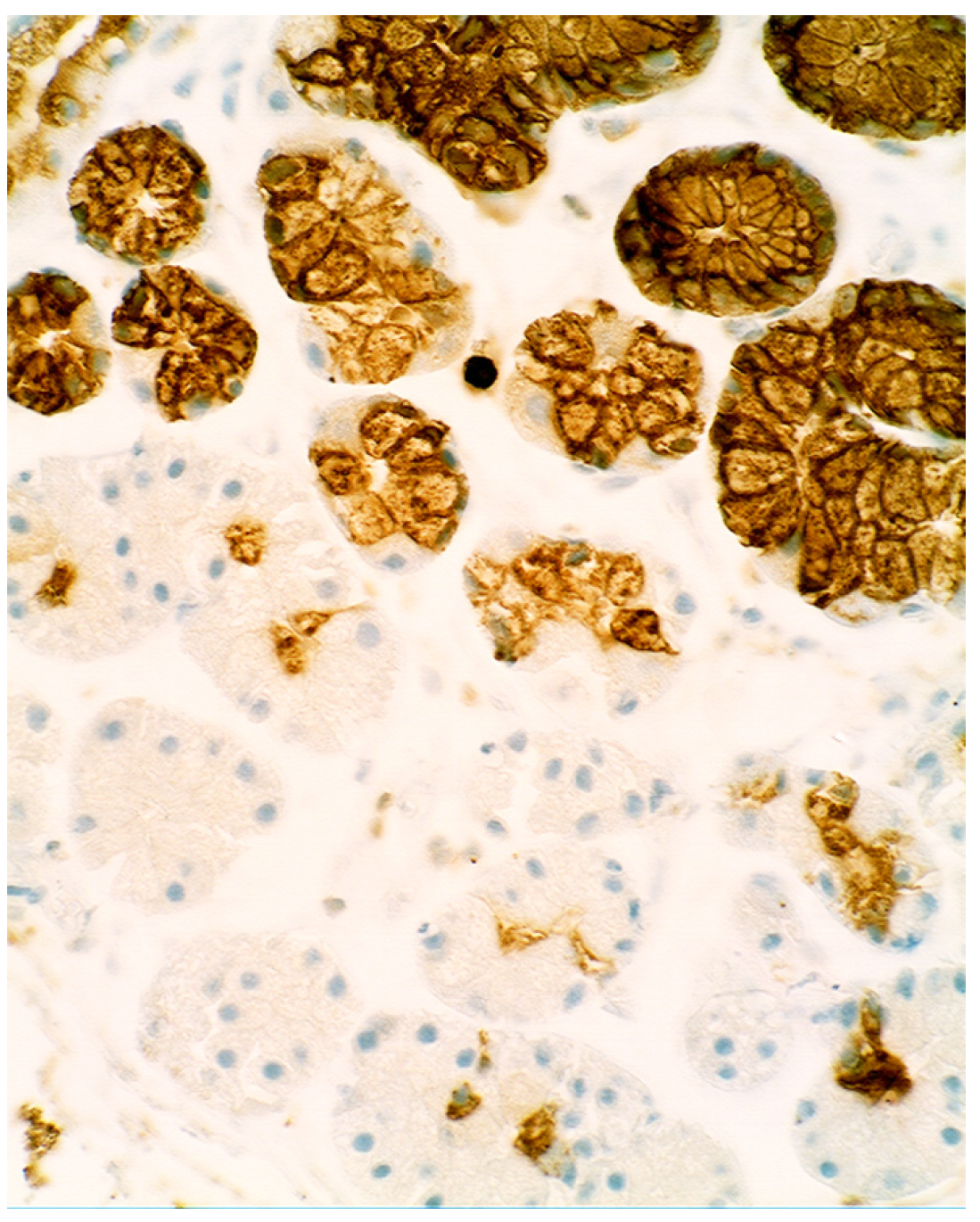
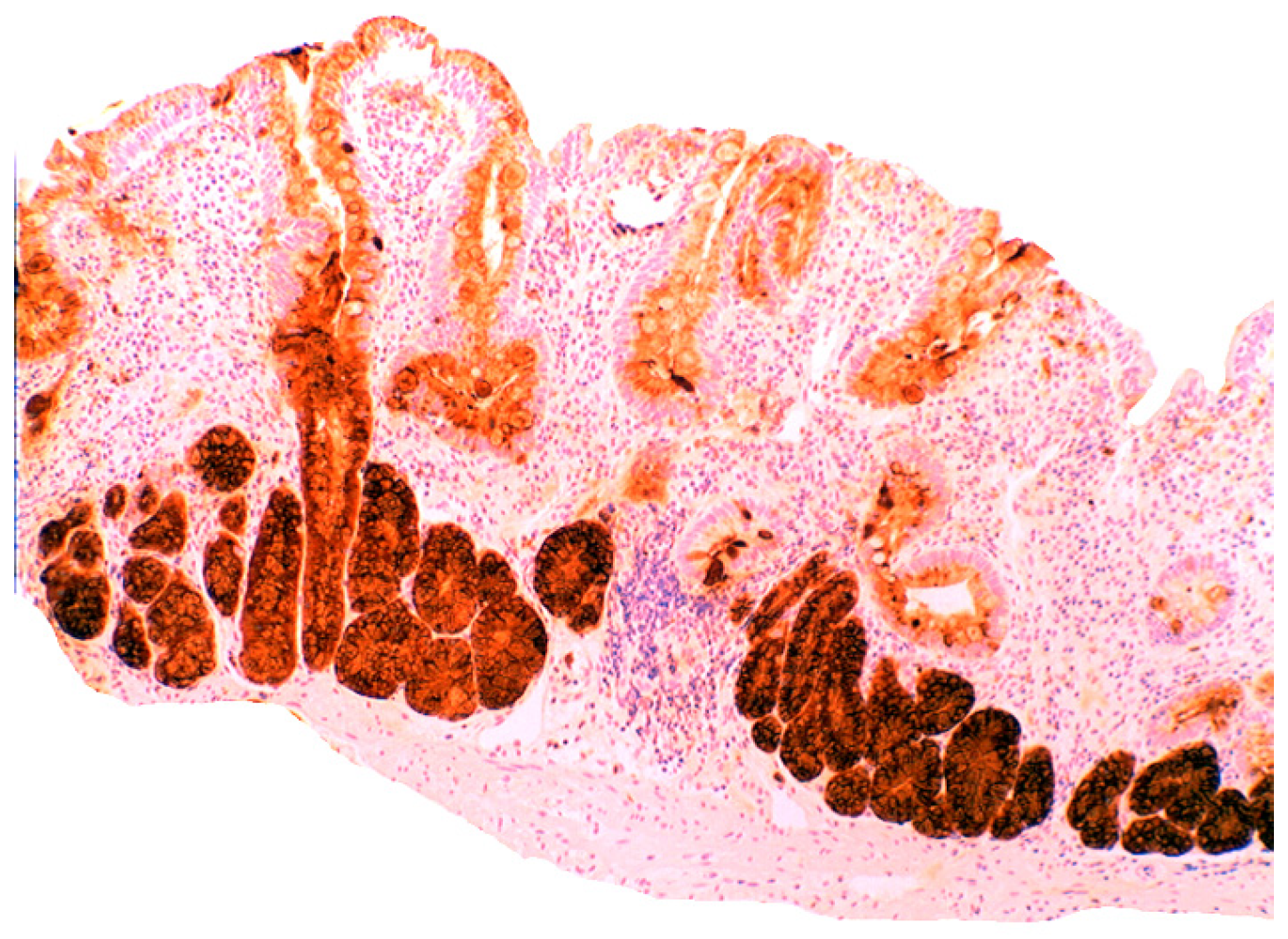
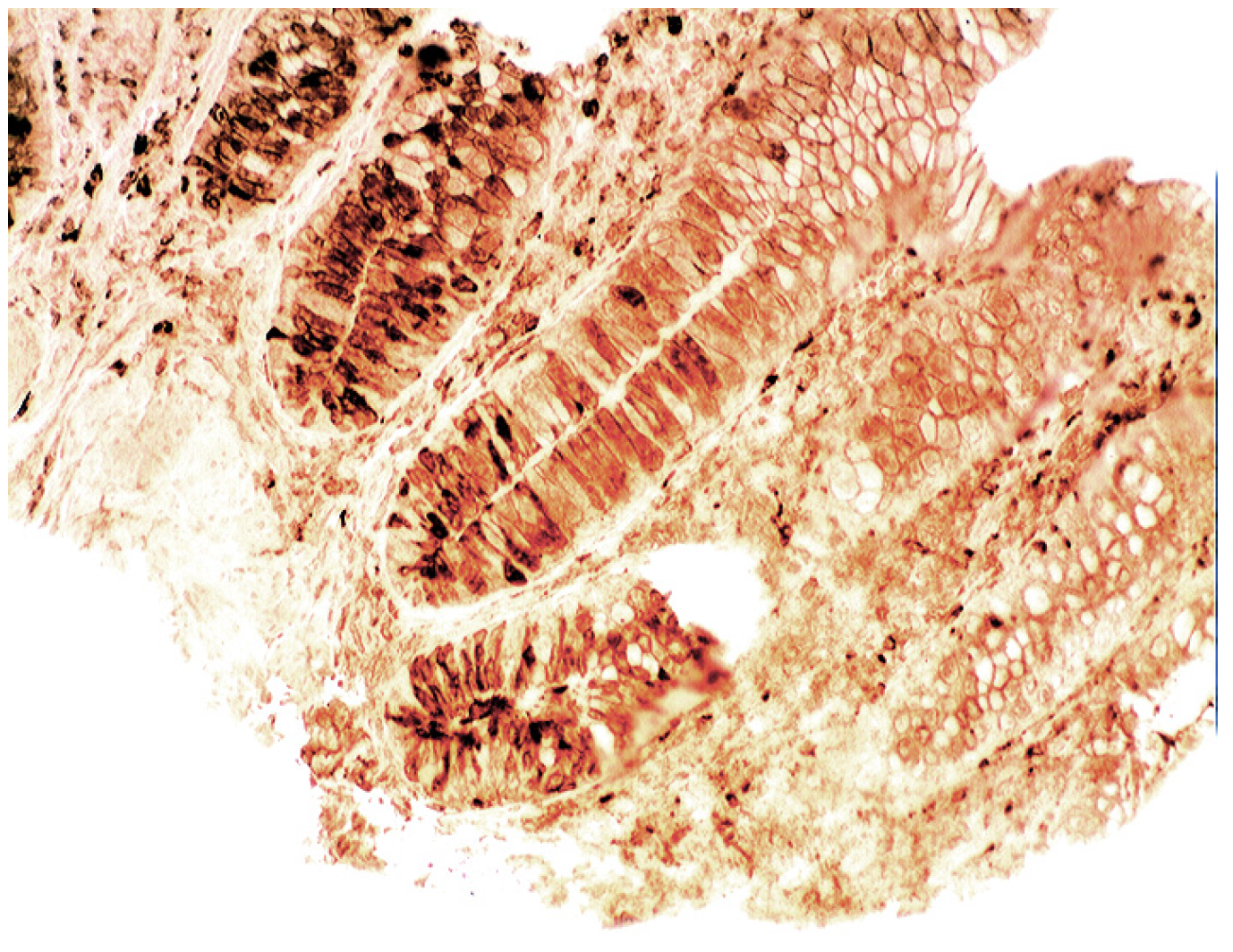
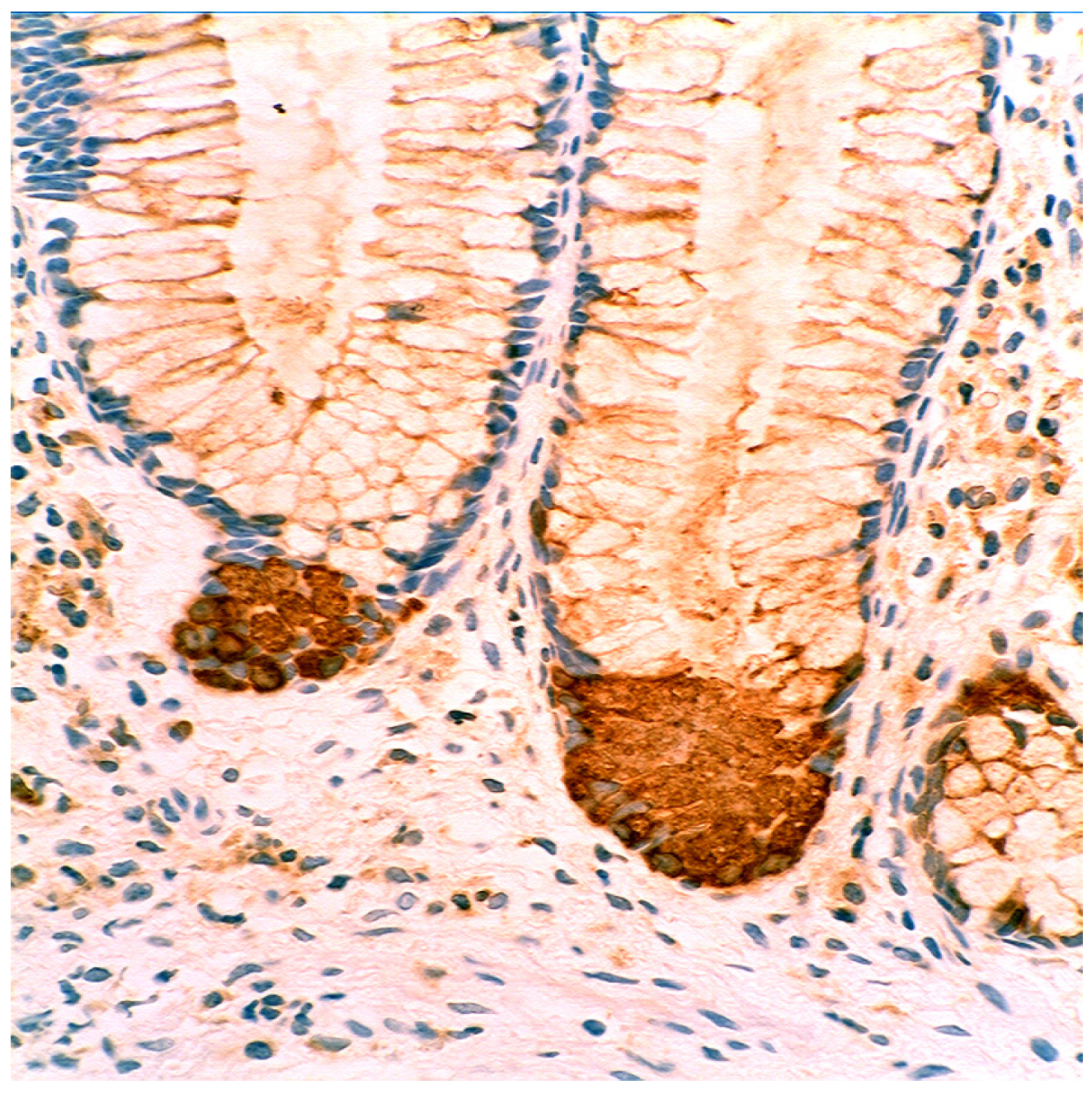
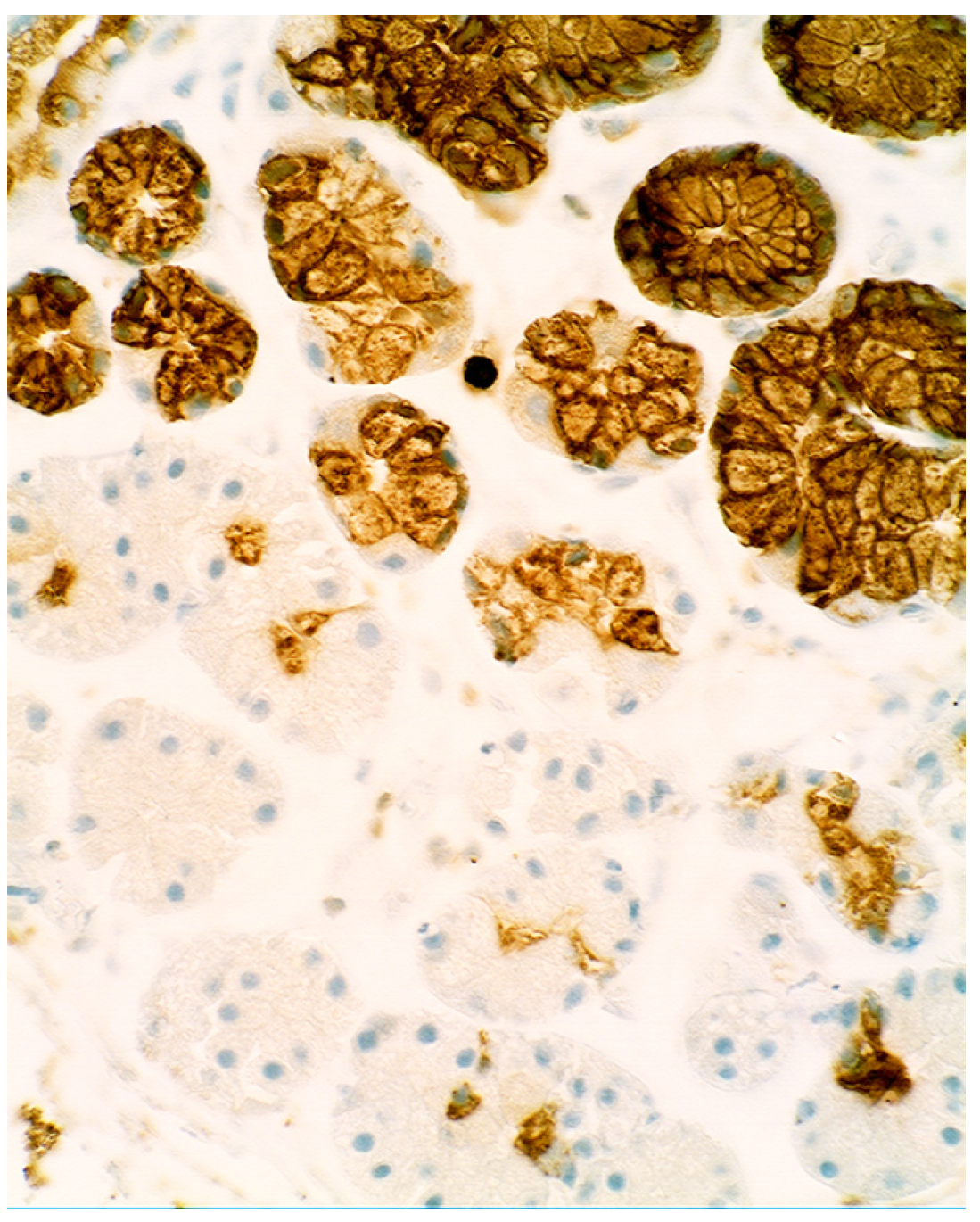
3.5. Lysozyme Is Up-Regulated in Chronic Gastritis, Intestinal Metaplasia, Autoimmune Gastritis and Fundic-Gland Polyps

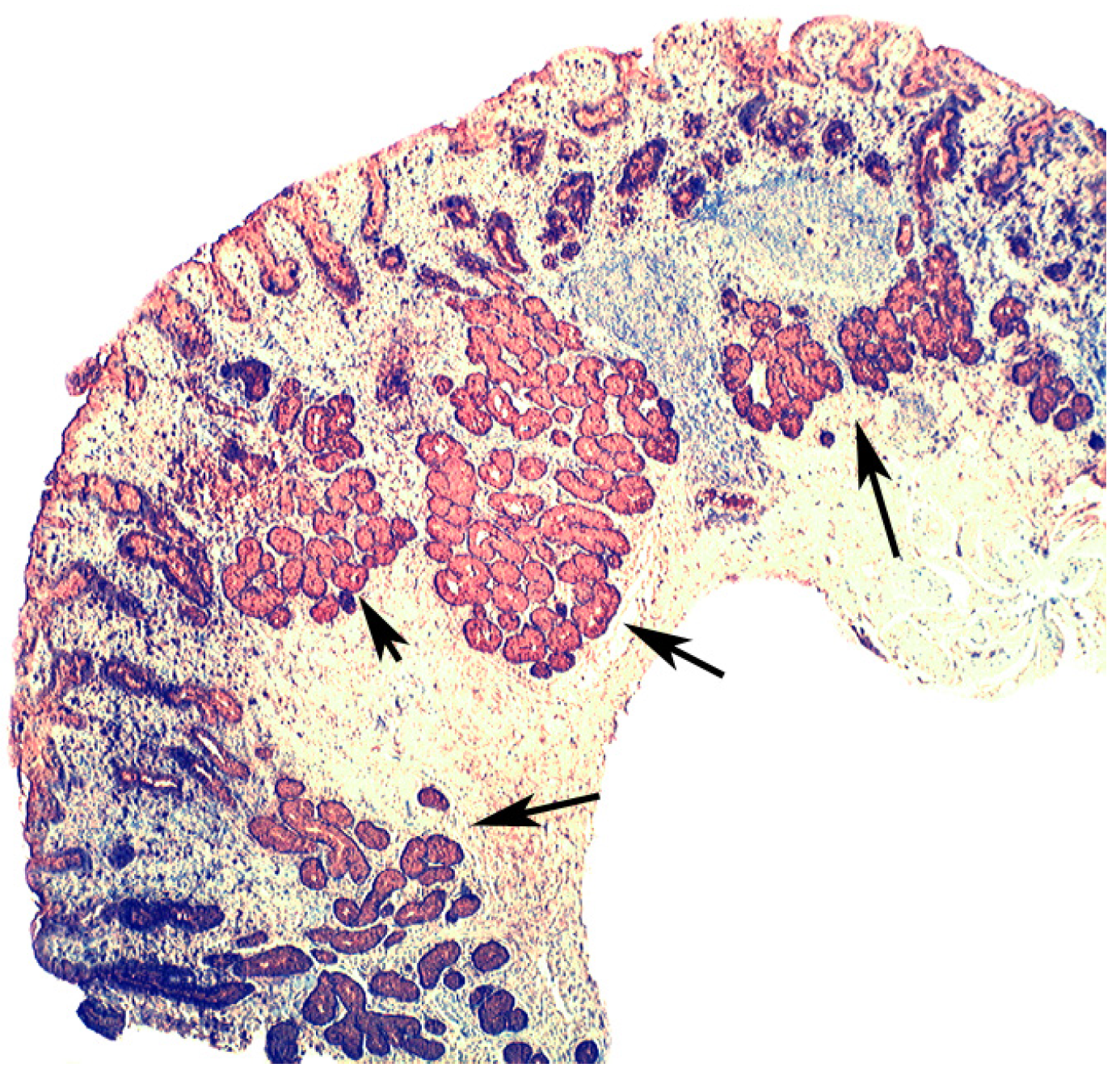
4. Intestinal Metaplasia in Disparate Geographical Regions
4.1. Fundic Gland Polyps
4.2. Absence of H. pylori in Fundic Gland Polyps
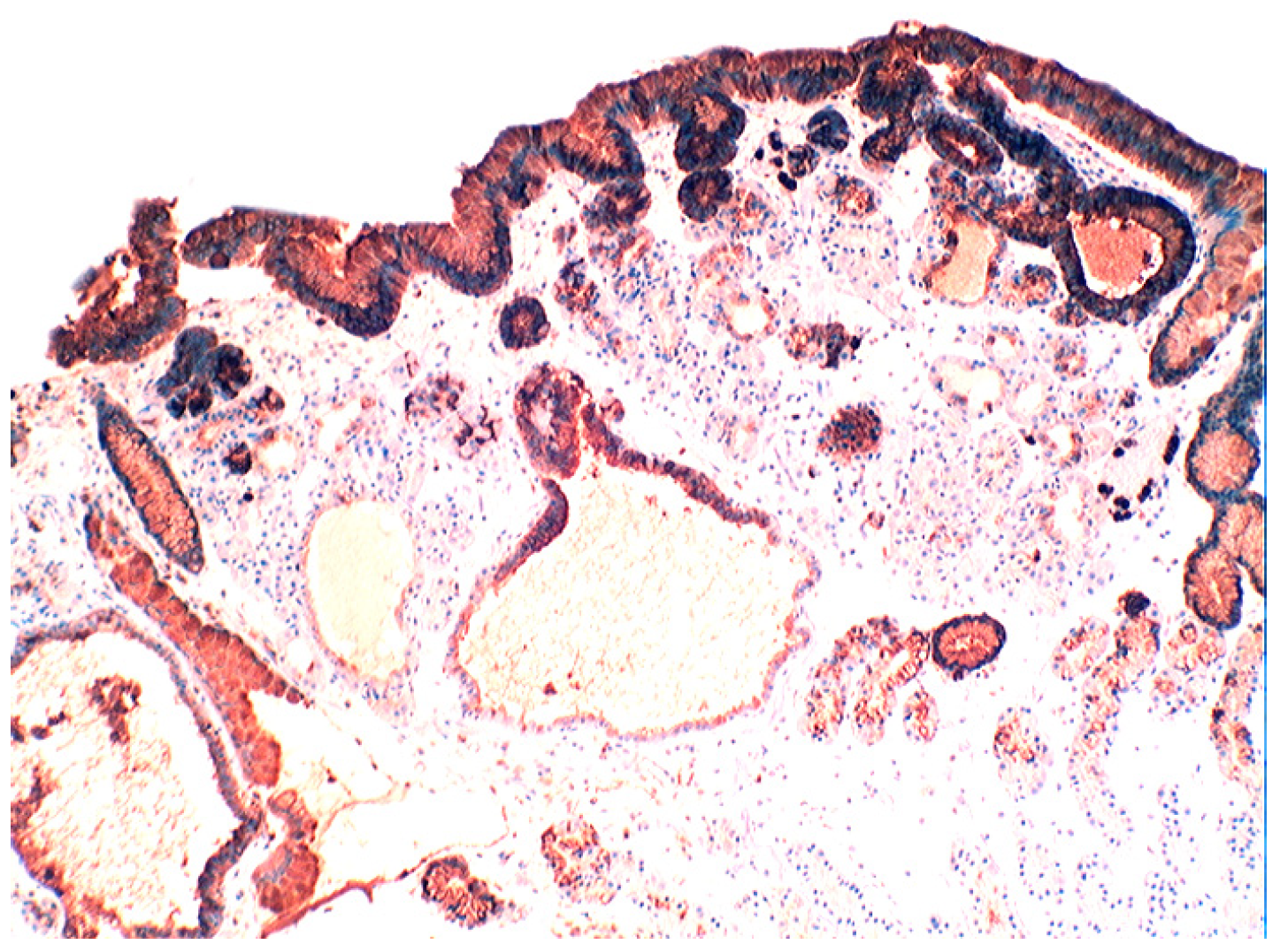
4.3. Coeliac Disease
4.4. Bacteria in Coeliac Disease
4.5. Lysozyme is Up-Regulated in Coeliac Disease
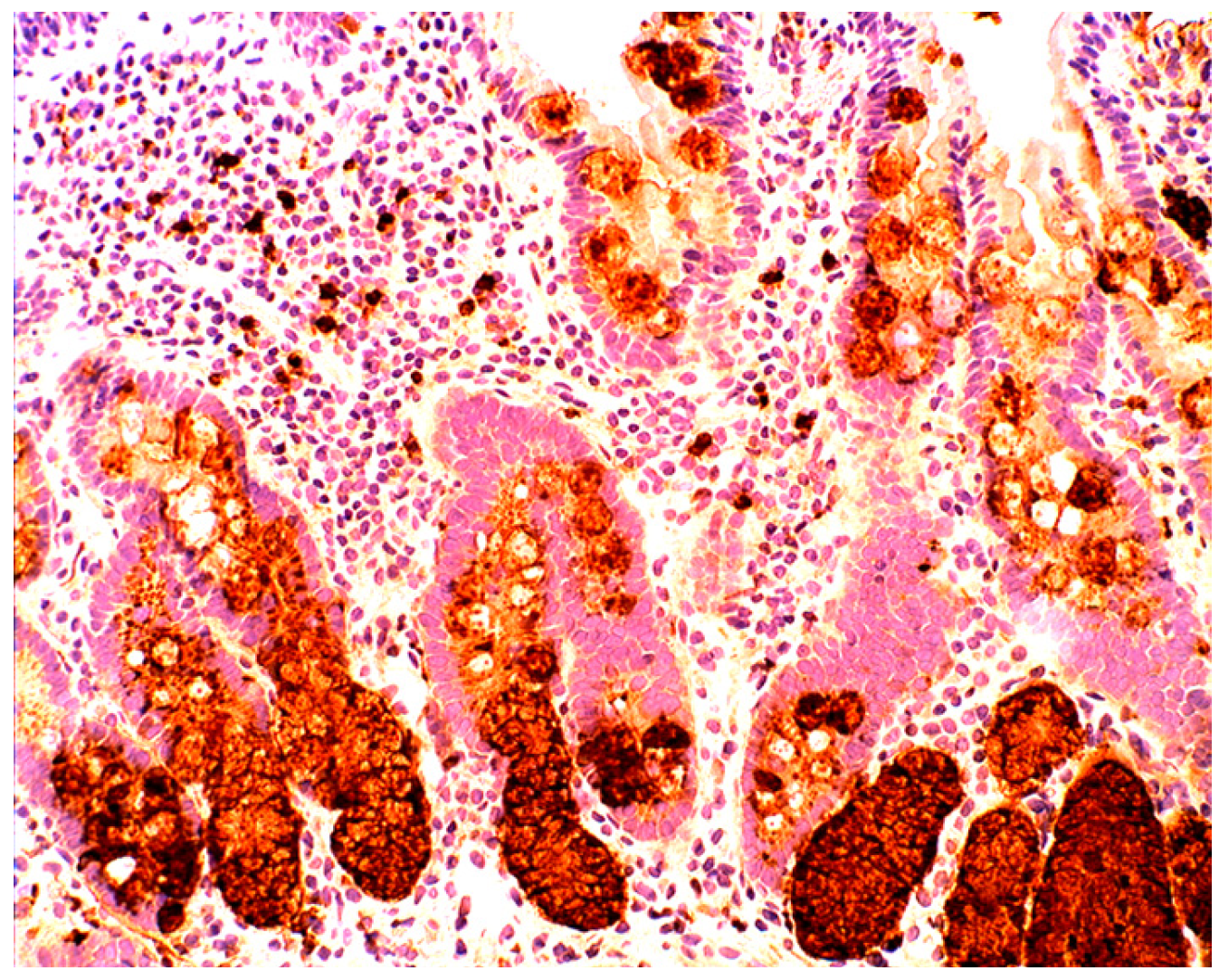
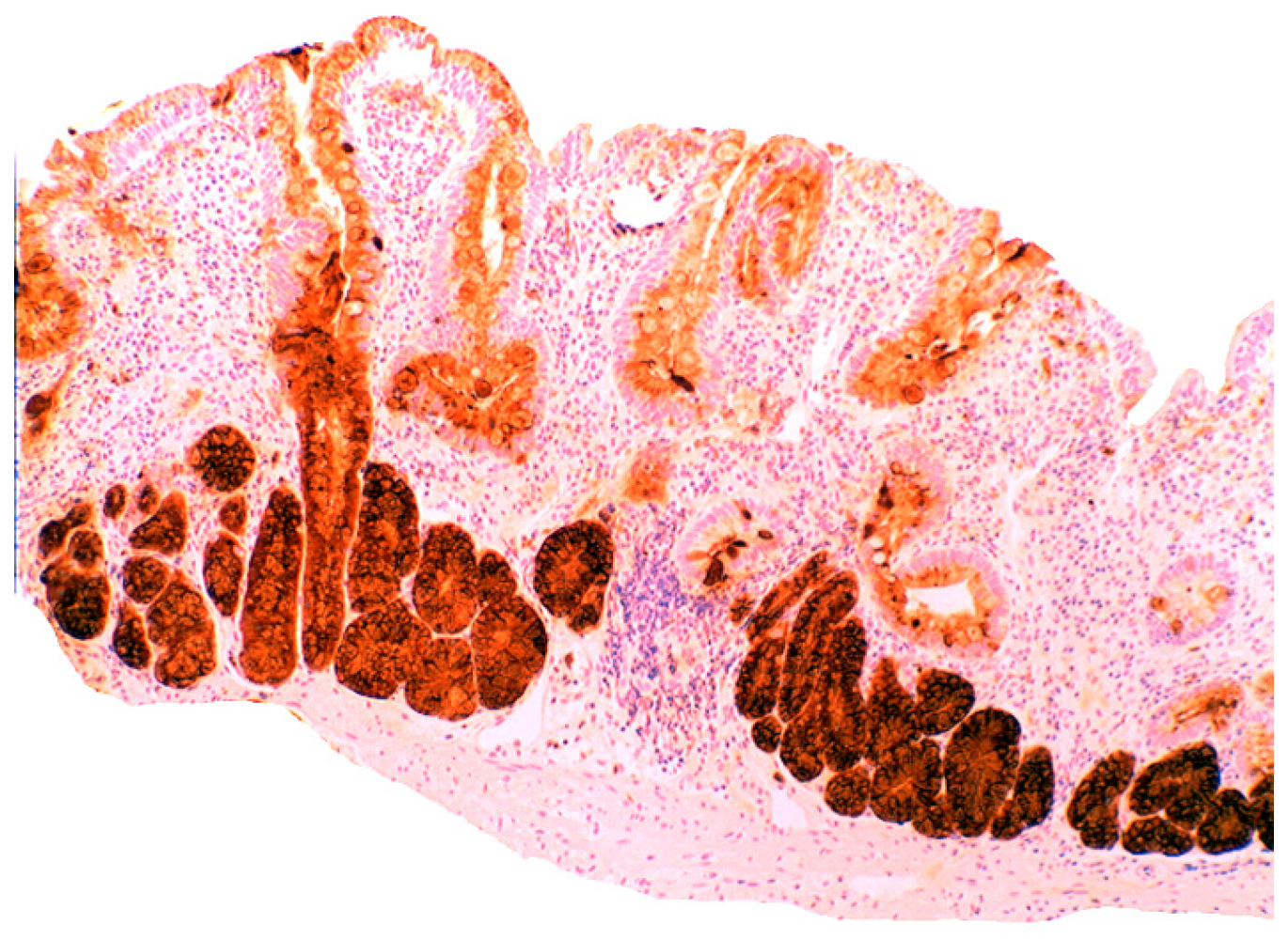
5. Collagenous Colitis, Lymphocytic Colitis, Ulcerative Colitis and Crohn’s Colitis
5.1. Bacteria in Microscopic Colitis
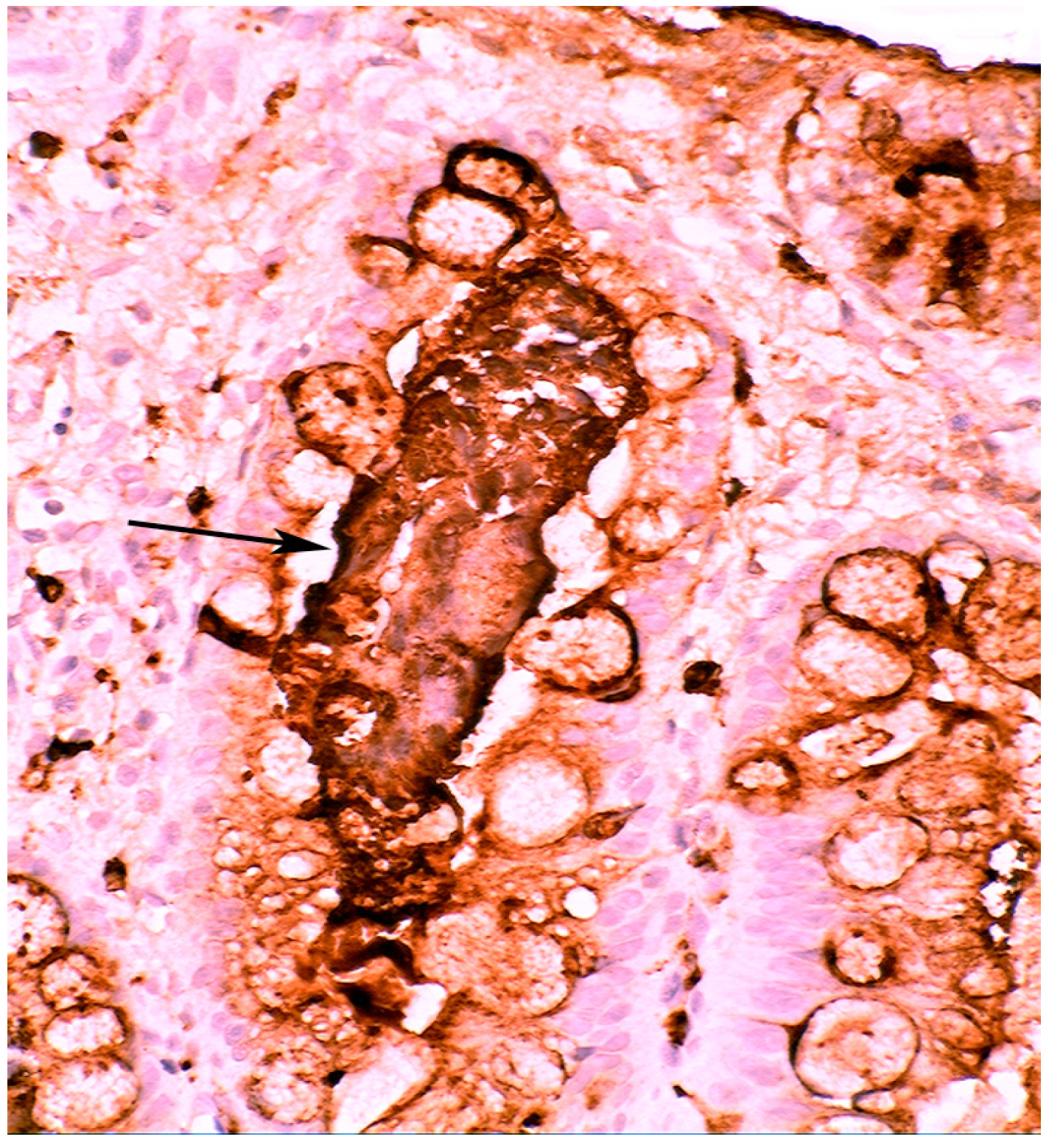
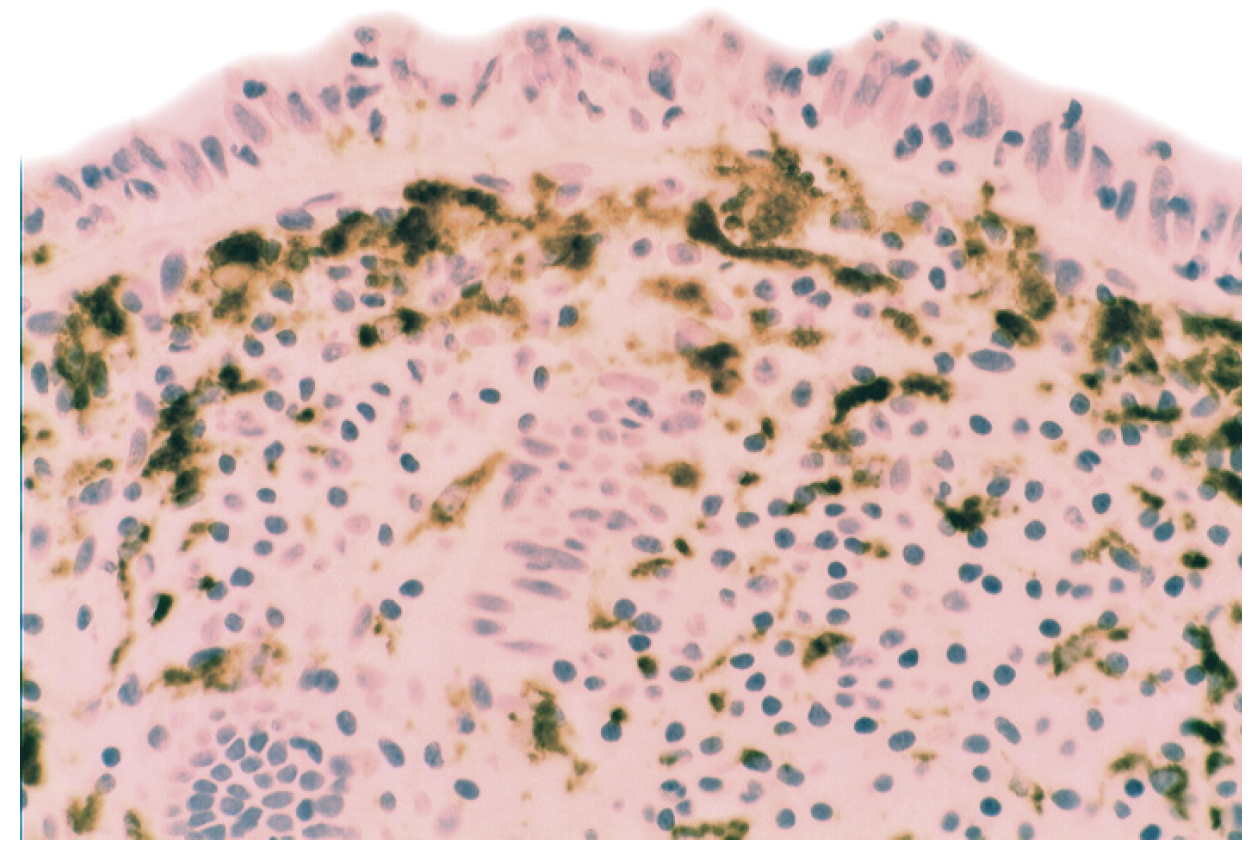
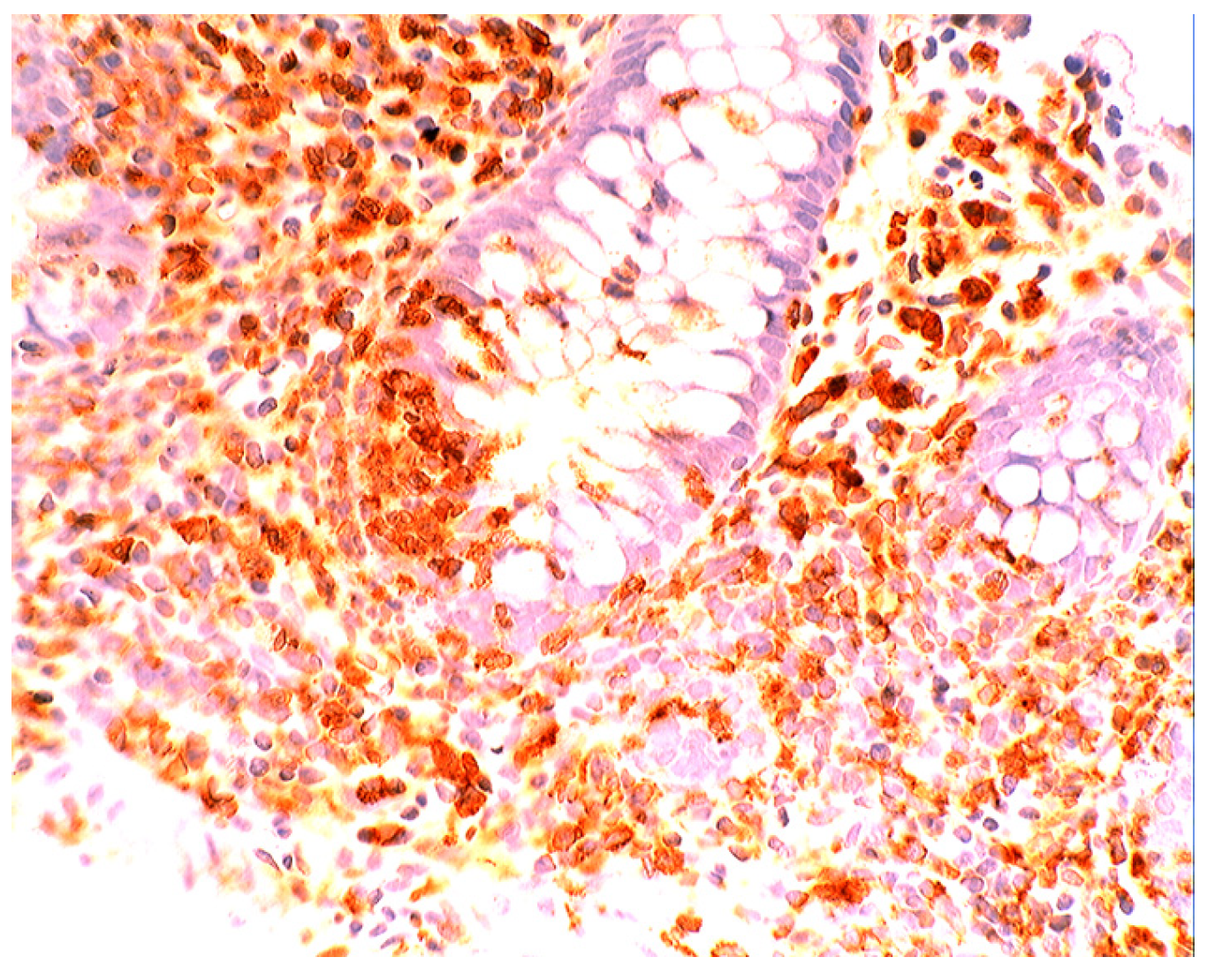
5.2. Lysozyme Is Up-Regulated in Microscopic Colitis
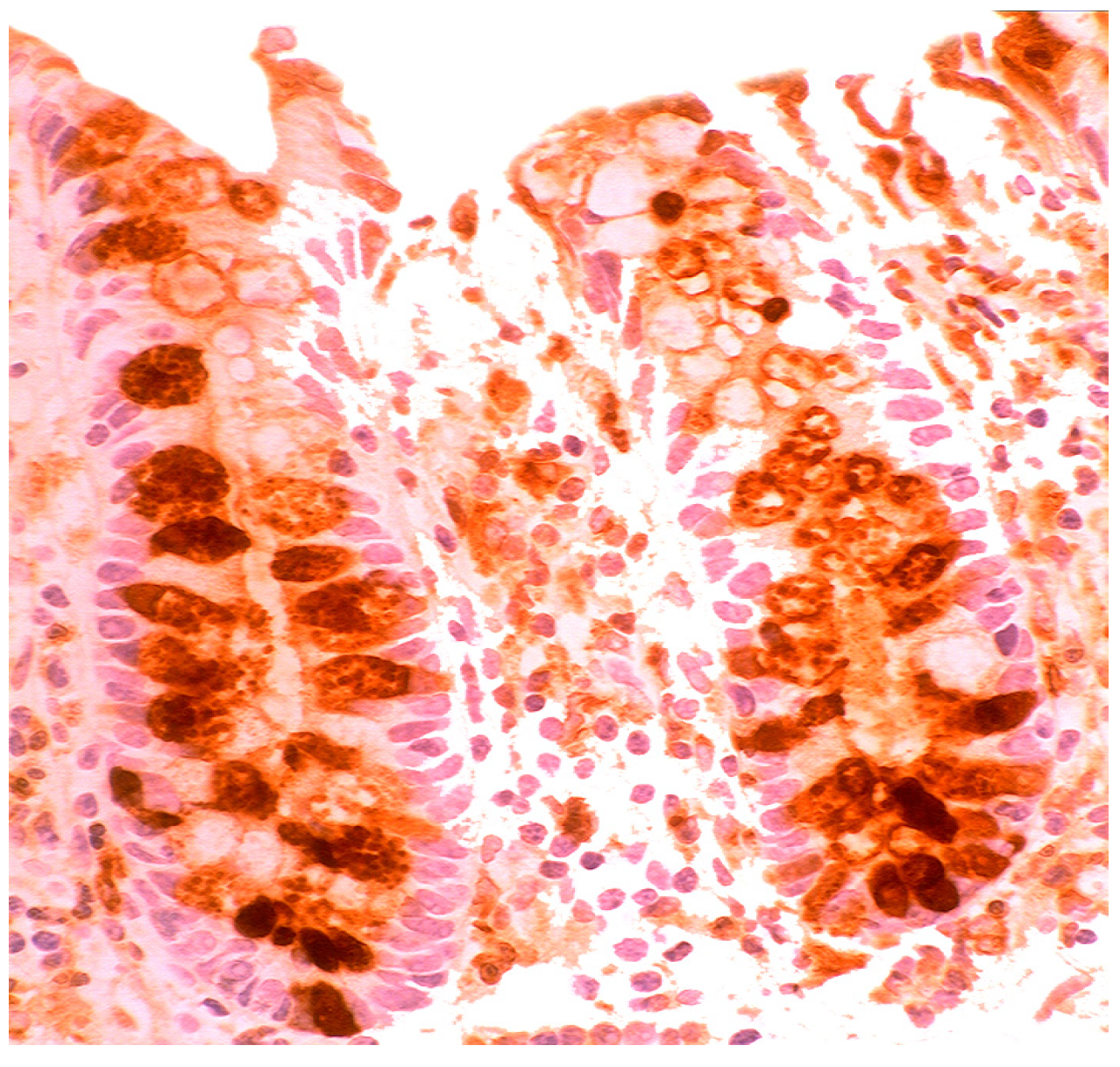
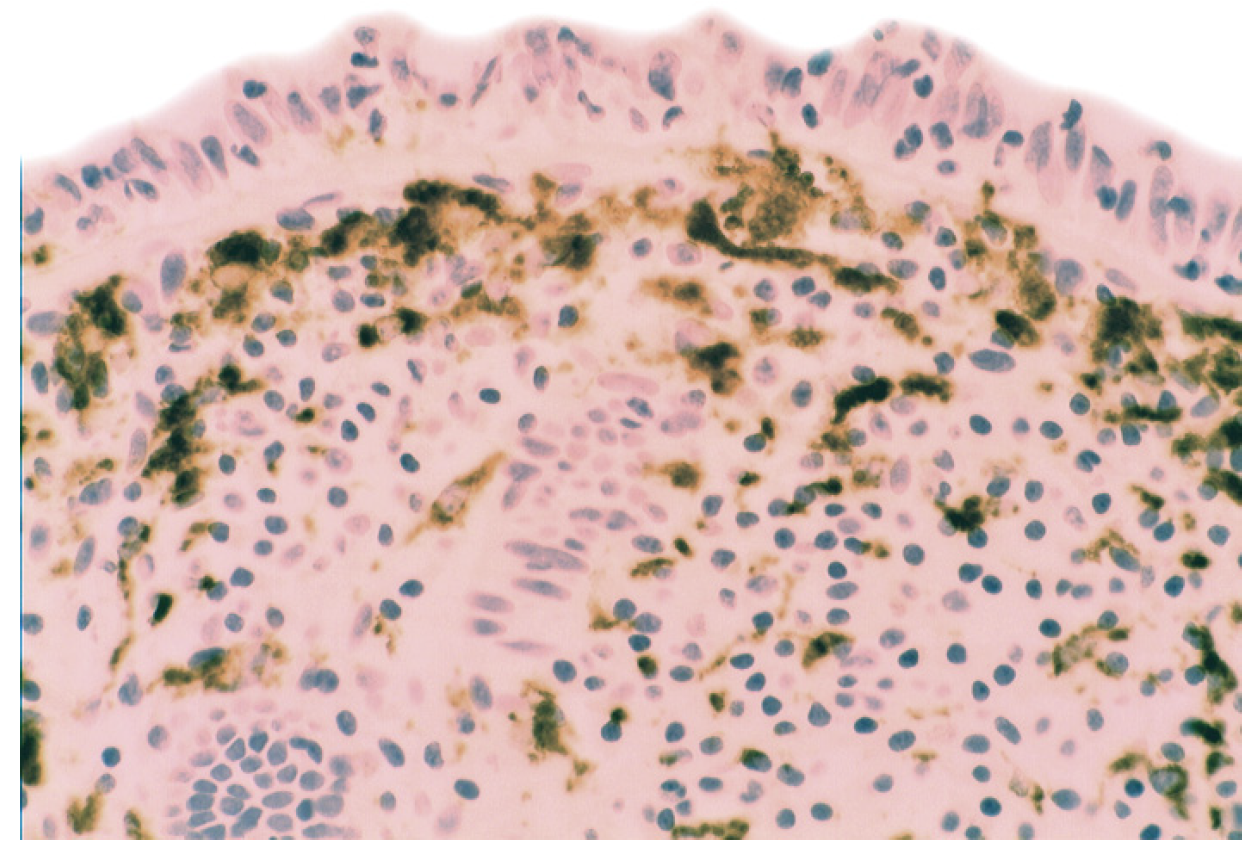
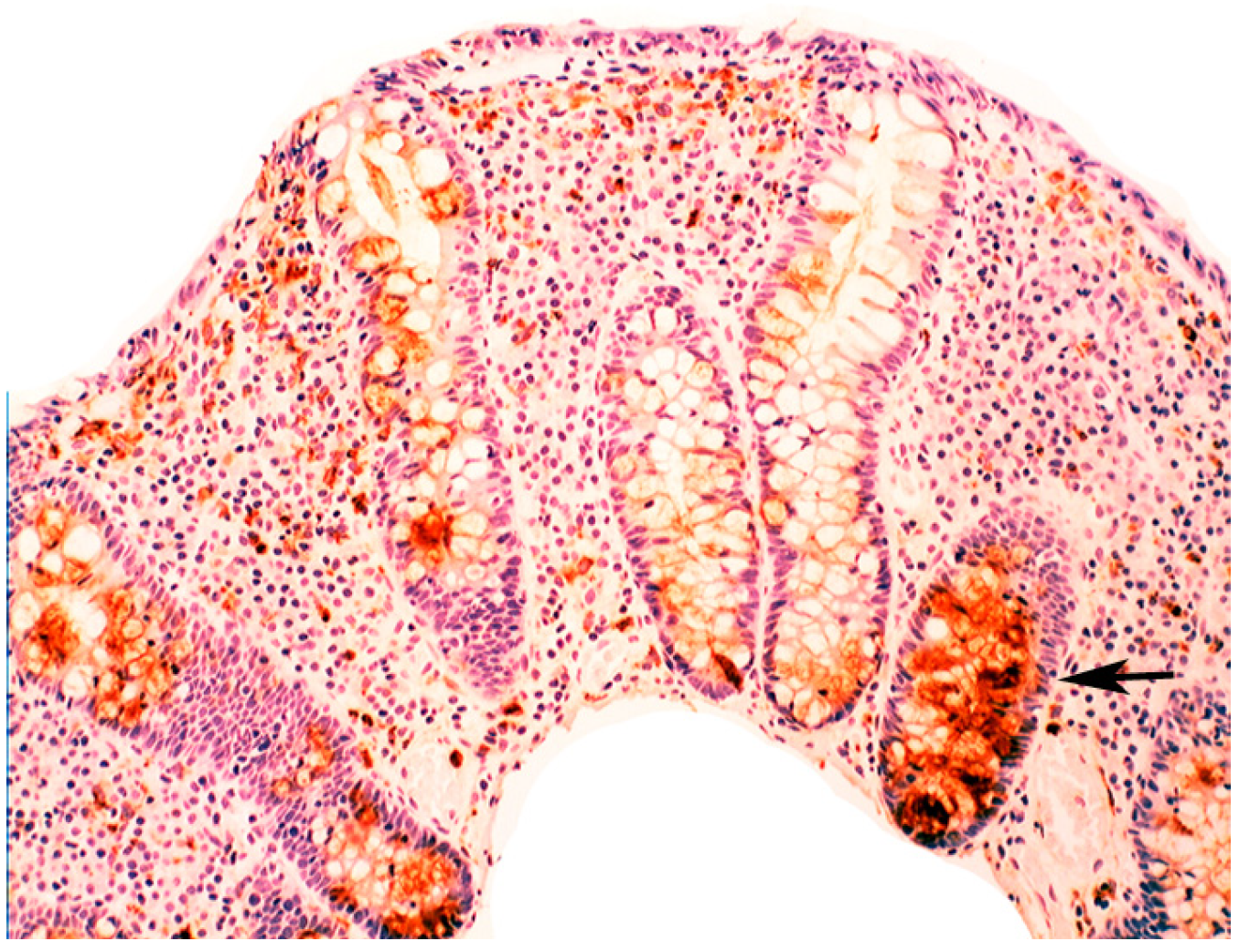
5.3. Bacteria in Inflammatory Bowel Disease
5.4. Lysozyme Up-Regulated in Inflammatory Bowel Disease
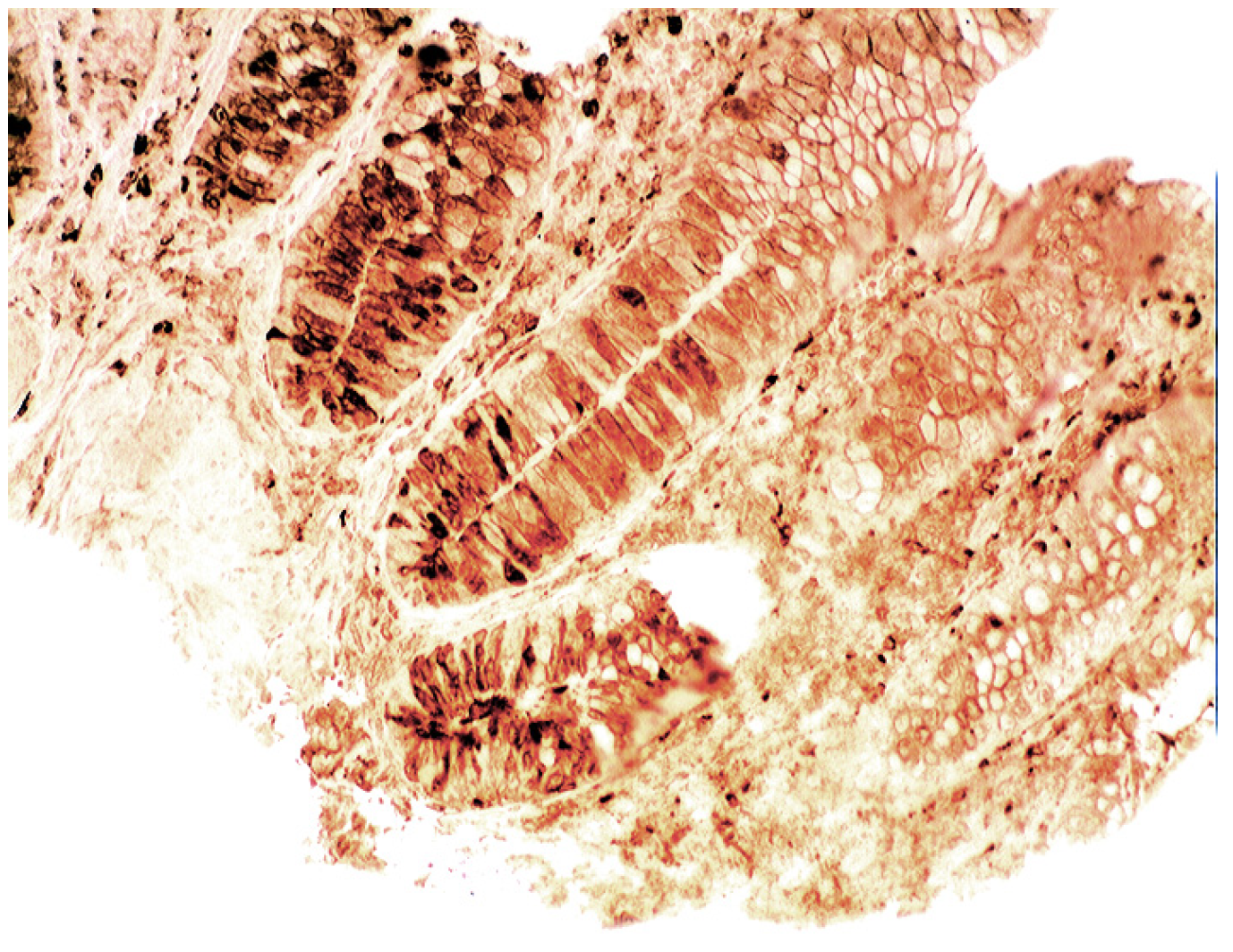

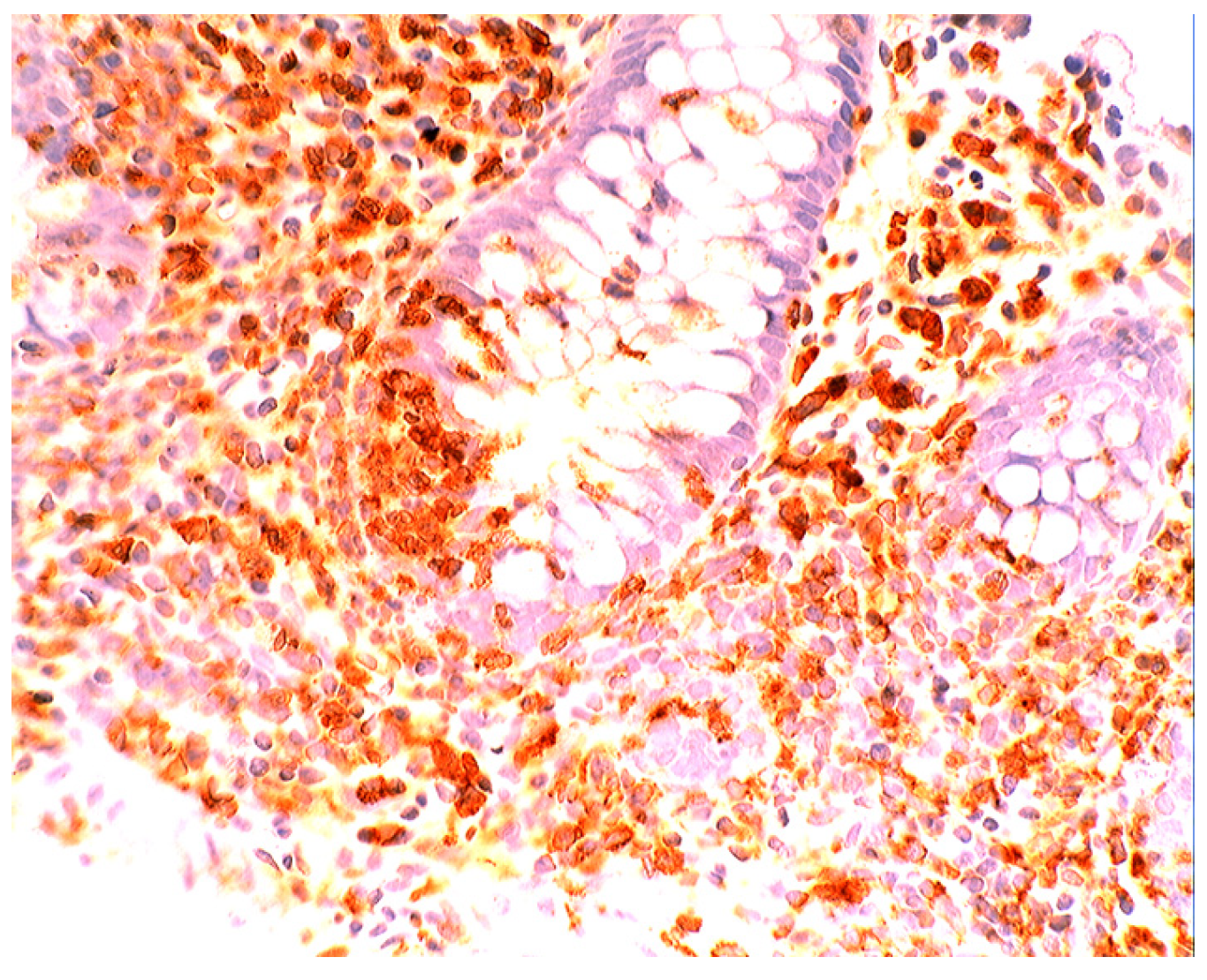
6. Epilogue
Conflicts of Interest
References
- Kaper, J.B.; Sperandio, V. Bacterial cell-to-cell signaling in the gastrointestinal tract. Infect. Immun. 2005, 73, 3197–3209. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Blaser, M.J.; Ley, R.E.; Knight, R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 2011, 140, 1713–1719. [Google Scholar] [CrossRef]
- Fleming, A. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. Sect. B 1922, 93, 306–312. [Google Scholar] [CrossRef]
- Yoshimura, K.; Toibana, A.; Nakahama, K. Human lysozyme: Sequencing of a cDNA, and expression and secretion by Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1988, 150, 794–801. [Google Scholar] [CrossRef]
- Peters, C.; Kruse, U.; Pollwein, R.; Grzeschik, K.; Sippel, T. The human lysozyme gene. Sequence organization and chromosomal localization. Eur. J. Biochem. 1989, 182, 507–512. [Google Scholar] [CrossRef]
- Sahoo, N.R.; Kumar, P.; Bhusan, B.; Bhattacharya, T.K.; Dayal, S.; Sahoo, M. Lysozyme in livestock: A guide to selection for disease resistance: A review. J. Anim. Sci. Adv. 2012, 2, 347–360. [Google Scholar]
- Rubio, C.A.; Lörinc, E. Lysozyme is up-regulated in Barrett’s mucosa. Histopathology 2011, 58, 796–799. [Google Scholar] [CrossRef]
- Rubio, C.A.; Befrits, R. Increased lysozyme expression in gastric biopsies with intestinal metaplasia and pseudopyloric metaplasia. Int. J. Clin. Exp. Med. 2009, 2, 248–253. [Google Scholar]
- Rubio, C.A. Lysozyme expression in microscopic colitis. J. Clin. Pathol. 2011, 64, 510–515. [Google Scholar] [CrossRef]
- Rubio, C.A. Lysozyme-rich mucus metaplasia in duodenal crypts supersedes Paneth cells in celiac disease. Virchows Arch. 2011, 459, 339–346. [Google Scholar] [CrossRef]
- Appelman, H.D.; Umar, A.; Orlando, R.C.; Sontag, S.J.; Nandurkar, S.; el-Zimaity, H.; Lanas, A.; Parise, P.; Lambert, R.; Shields, H.M. Barrett’s esophagus: Natural history. Ann. N. Y. Acad. Sci. 2011, 1232, 292–308. [Google Scholar] [CrossRef]
- Rubio, C.A.; Dick, E.J.; Schlabritz-Loutsevitch, N.E.; Orrego, A.; Hubbard, G.B. The columnar-lined mucosa at the gastroesophageal junction in non-human primates. Int. J. Clin. Exp. Pathol. 2009, 2, 481–488. [Google Scholar]
- Rubio, C.A.; Nilsson, J.R.; Owston, M.; Dick, E.J., Jr. The length of the Barrett's mucosa in baboons, revisited. Anticancer Res. 2012, 32, 3115–3118. [Google Scholar]
- Spechler, S.; Goyal, R.K. Barrett’s esophagus. N. Engl. J. Med. 1986, 315, 362–367. [Google Scholar] [CrossRef]
- Sampliner, R.E. Practice guidelines on the diagnosis, surveillance, and therapy of Barrett’s esophagus. The Practice Parameters Committee of the American College of Gastroenterology. Am. J. Gastroenterol. 1998, 93, 1028–1032. [Google Scholar] [CrossRef]
- Playford, R.J. New British Society of Gastroenterology (BSG) guidelines for the diagnosis and management of Barrett’s oesophagus. Gut 2006, 55, 442–444. [Google Scholar] [CrossRef]
- Fiocca, R.; Mastracci, L.; Milione, M.; Parente, P.; Savarino, V. Gruppo Italiano Patologi Apparato Digerente (GIPAD) and Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP). Microscopic esophagitis and Barrett’s esophagus: The histology report. Dig. Liver Dis. 2011, 43, S319–S330. [Google Scholar] [CrossRef]
- Takubo, K.; Vieth, M.; Aida, J.; Sawabe, M.; Kumagai, Y.; Hoshihara, Y.; Arai, T. Differences in the definitions used for esophageal and gastric diseases in different countries: Endoscopic definition of the esophagogastric junction, the precursor of Barrett’s adenocarcinoma, the definition of Barrett’s esophagus, and histologic criteria for mucosal adenocarcinoma or high-grade dysplasia. Digestion 2009, 80, 248–257. [Google Scholar] [CrossRef]
- Waldum, H.L.; Hauso, Ø.; Sandvik, A.K. PPI-induced hypergastrinaemia and Barrett’s mucosa: The fog thickens. Gut 2010, 59, 1157–1158. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z.; Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; et al. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef]
- Pei, Z.; Yang, L.; Peek, R.M., Jr.; Levine, S.M.; Pride, D.T.; Blaser, M.J. Bacterial biota in reflux esophagitis and Barrett’s esophagus. World J. Gastroenterol. 2005, 11, 7277–7283. [Google Scholar]
- Macfarlane, S.; Furrie, E.; Macfarlane, G.; Dillon, J. Microbial colonization of the upper gastrointestinal tract in patients with Barrett’s esophagus. Clin. Infect. Dis. 2007, 45, 29–38. [Google Scholar] [CrossRef]
- Liu, N.; Ando, T.; Ishiguro, K.; Maeda, O.; Watanabe, O.; Funasaka, K.; Nakamura, M.; Miyahara, R.; Ohmiya, N.; Goto, H. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett’s esophagus. BMC Infect. Dis. 2013, 13, 130–136. [Google Scholar] [CrossRef]
- Rubio, C.A. Lysozyme is up-regulated in columnar-lined Barrett’s mucosa: A possible natural defence mechanism against Barrett’s esophagus-associated pathogenic bacteria. Anticancer Res. 2012, 32, 5115–5119. [Google Scholar]
- Sarosi, G.; Brown, G.; Jaiswal, K.; Feagins, L.A.; Lee, E.; Crook, T.W.; Souza, R.F.; Zou, Y.S.; Shay, J.W.; Spechler, S.J. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis. Esophagus 2008, 21, 43–50. [Google Scholar]
- Rubio, C.A. Putative Stem Cells in Mucosas of the Esophago-Gastrointestinal Tract. Chapter 10; In Stem Cell, Regenerative Medicine and Cancer; Singh, S.R., Ed.; Nova Science Publishers, Inc.: Haupauge, NY, USA, 2011; pp. 281–310. [Google Scholar]
- Misiewicz, J.; Tygat, G.; Goodwin, C. The Sydney System: A new classification of gastritis. Work. Party Rep. 1990, 1, 10–11. [Google Scholar]
- Cruveihier, J. Traté d´anatomie pathologique genéràle; Bulletins de la Société d'anthropologie de Paris, Ed.; Billièr: Paris, France, 1862; p. 48. [Google Scholar]
- MacDonald, W.; Rubin, C. Gastric biopsy: A critical evaluation. Gastroenterology 1967, 53, 143–170. [Google Scholar]
- Edwards, F.; Coghill, N. Aetiological factors in chronic atrophic gastritis. Br. Med. J. 1966, ii, 1400–1415. [Google Scholar]
- Warren, J.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, I, 1273–1275. [Google Scholar]
- Correa, P. A human model of gastric carcinogenesis. Cancer Res. 1988, 48, 1319–1326. [Google Scholar]
- Oh, J.D.; Kling-Backhed, H.; Giannakis, M.; Engstrand, L.G.; Gordon, J.I. Interactions between gastric epithelial stem cells and Helicobacter pylori in the setting of chronic atrophic gastritis. Curr. Opin. Microbiol. 2006, 9, 21–27. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis: The updated Sydney System. International workshop on the histopathology of gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar]
- Giannella, R.; Broitman, S.; Zamcheck, N. Gastric acid barrier to ingested micro-organisms in man: Studies in vivo and in vitro. Gut 1972, 13, 251–256. [Google Scholar] [CrossRef]
- Rubio, C.A.; Jaramillo, E.; Suzuki, G.; Lagergren, P.; Nesi, G. Antralization of the gastric mucosa of the incisura angularis an its gastrin expression. Int. J. Clin. Exp. Pathol. 2009, 2, 65–70. [Google Scholar]
- Nakajima, S.; Nishiyama, Y.; Yamaoka, M.; Yasuoka, T.; Cho, E. Changes in the prevalence of Helicobacter pylori infection and gastrointestinal diseases in the past 17 years. J. Gastroenterol. Hepatol. 2010, 25, S99–S110. [Google Scholar] [CrossRef]
- Campbell, D.; Warren, B.; Thomas, J.; Figura, N.; Telford, J.; Sullivan, P. The African enigma: Low prevalence of gastric atrophy, high prevalence of chronic inflammation in West African adults and children. Helicobacter 2001, 6, 263–267. [Google Scholar] [CrossRef]
- Graham, D.Y.; Lu, H.; Yamaoka, Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: Facts or medical myths. J. Dig. Dis. 2009, 10, 77–84. [Google Scholar] [CrossRef]
- Saieva, C.; Rubio, C.A.; Nesi, G.; Zini, E.; Filomena, A. Classification of gastritis in first-degree relatives of patients with gastric cancer in a high cancer-risk area in Italy. Anticancer Res. 2012, 32, 1711–1716. [Google Scholar]
- Torbenson, M.; Abraham, S.C.; Boitnott, J.; Yardley, J.H.; Wu, T. Autoimmune gastritis: Distinct histological and immuno-histochemical findings before complete loss of oxyntic glands. Mod. Pathol. 2002, 15, 102–109. [Google Scholar] [CrossRef]
- Petersson, F.; Borch, K.; Franzén, E. Prevalence of subtypes of intestinal metaplasia in the general population and in patients with autoimmune chronic atrophic gastritis. Scand. J. Gastroenterol. 2002, 37, 262–266. [Google Scholar] [CrossRef]
- Rubio, C.A.; Kaufeldt, A. Paucity of synaptophysin-expressing cells in Barrett’s mucosa. Histopathology 2013, 63, 208–216. [Google Scholar] [CrossRef]
- Guerre, J.; Vedel, G.; Gaudric, M.; Paul, G.; Cornuau, J. Bacterial flora in gastric juice taken at endoscopy in 93 normal subjects. Pathol. Biol. 1986, 34, 57–60. [Google Scholar]
- Monstein, H.J.; Tiveljung, A.; Kraft, C.H.; Borch, K.; Jonasson, J. Profiling of bacterial flora in gastric biopsies from patients with Helicobacter pylori associated gastritis and histologically normal control individuals by temperature gradient gel electrophoresis and 16S rDNA sequence analysis. J. Med. Microbiol. 2000, 49, 817–822. [Google Scholar]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. USA 2006, 103, 732–737. [Google Scholar]
- Li, X.X.; Wong, G.L.; To, K.F.; Wong, V.W.; Lai, L.H. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-Inflammatory drug use. PLoS ONE 2009, 4, e7985. [Google Scholar]
- Shen, B.; Porter, E.M.; Reynoso, E.; Shen, C.; Ghosh, D.; Connor, J.T.; Drazba, J.; Rho, H.K.; Gramlich, T.L.; Li, R.; et al. Human defensin 5 expression in intestinal metaplasia of the upper gastrointestinal tract. J. Clin. Pathol. 2005, 58, 687–691. [Google Scholar] [CrossRef]
- Rubio, C.A. My approach to reporting a gastric biopsy. J. Clin. Pathol. 2007, 60, 160–166. [Google Scholar] [CrossRef]
- Shousha, S.; el-Sherif, A.M.; el-Guneid, A.; Arnaout, A.H. Helicobacter pylori and intestinal metaplasia: Comparison between British and Yemeni patients. Am. J. Gastroenterol. 1993, 88, 1373–1376. [Google Scholar]
- Rubio, C.A.; Kato, Y.; Sugano, H.; Kitagawa, T. Intestinal metaplasia of the stomach in Swedish and Japanese patients without ulcers or carcinoma. Jpn. J. Cancer Res. 1987, 78, 467–472. [Google Scholar]
- Rubio, C.A.; Jessurum, J.; Kato, Y. Low frequency of intestinal metaplasia in gastric biopsies from Mexican patients: A comparison with Japanese and Swedish patients. Jpn. J. Cancer Res. 1992, 83, 491–494. [Google Scholar] [CrossRef]
- Shand, A.; Taylor, A.; Banerjee, M.; Lessels, A.; Coia, J.; Clark, C.; Haites, N.; Ghosh, S. Gastric fundic gland polyps in south-east Scotland: Absence of adenomatous polyposis coli gene mutations and a strikingly low prevalence of Helicobacter pylori infection. J. Gastroenterol. Hepatol. 2002, 17, 1161–1164. [Google Scholar] [CrossRef]
- Rubio, C.A. Plugs clog the glandular outlets in fundic gland polyps. Int. J. Clin. Exp. Pathol. 2009, 3, 69–74. [Google Scholar]
- Rubio, C.A. Lysozyme overexpression in fundic gland polyps. Anticancer Res. 2010, 30, 1021–1024. [Google Scholar]
- Dewar, D.H.; Ciclitira, P.J. Clinical features and diagnosis of celiac disease. Gastroenterology 2005, 128, S19–S24. [Google Scholar] [CrossRef]
- Ivarsson, A.; Högberg, L.; Stenhammar, L.; Swedish Childhood Coeliac Disease Working Group. The swedish childhood coeliac disease working group after 20 years: History and future. Acta Paediatr. 2010, 99, 1429–1431. [Google Scholar] [CrossRef]
- Myléus, A.; Ivarsson, A.; Webb, C.; Danielsson, L.; Hernell, O.; Högberg, L.; Karlsson, E.; Lagerqvist, C.; Norström, F.; Rosén, A.; et al. Celiac disease revealed in 3% of Swedish 12-year-olds born during an epidemic. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 170–176. [Google Scholar] [CrossRef]
- Sánchez, E.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal Bacteroides species associated with coeliac disease. J. Clin. Pathol. 2010, 63, 1105–1111. [Google Scholar] [CrossRef]
- Schippa, S.; Lebba, V.; Barbato, M.; di Nardo, G.; Totino, V.; Checchi, M.P.; Longhi, C.; Maiella, G.; Cucchiara, S.; Conte, M.P. A distinctive “microbial signature” in celiac pediatric patients. BMC Microbiol. 2010, 10, 175–179. [Google Scholar] [CrossRef]
- Forsberg, G.; Fahlgren, A.; Hörstedt, P.; Hammarström, S.; Hernell, O.; Hammarström, M.L. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am. J. Gastroenterol. 2004, 99, 894–904. [Google Scholar] [CrossRef]
- Ou, G.; Hedberg, M.; Hörstedt, P.; Baranov, V.; Forsberg, G.; Drobni, M.; Sandström, O.; Hammarström, S. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am. J. Gastroenterol. 2009, 104, 3058–3067. [Google Scholar] [CrossRef]
- Rubio, C.A. Signaling pathways, gene regulation and duodenal neoplasias. Chapter 6; In Signaling, Gene Regulation and Cancer; Singh, S.R., Ed.; Nova Science Publishers, Inc.: Haupauge, NY, USA, 2013; pp. 83–110. [Google Scholar]
- Lindström, C.G. “Collagenous colitis” with watery diarrhoea: A new entity? Pathol Eur. 1976, 11, 87–89. [Google Scholar]
- Giardiello, F.M.; Lazenby, A.J.; Bayless, T.M.; Levine, E.J.; Bias, W.B.; Ladenson, P.W.; Yardley, J.H. Lymphocytic (microscopic) colitis. Clinico-pathologic study of 18 patients and comparison to collagenous colitis. Dig. Dis. Sci. 1989, 34, 1730–1738. [Google Scholar] [CrossRef]
- Wickbom, A.; Bohr, J.; Eriksson, S.; Udumyan, R.; Nyhlin, N.; Tysk, C. Stable incidence of collagenous colitis and lymphocytic colitis in Örebro, Sweden, 1999–2008: A continuous epidemiologic study. Inflamm. Bowel Dis. 2013, 19, 2387–2393. [Google Scholar] [CrossRef]
- Gustafsson, R.J.; Ohlsson, B.; Benoni, C.; Jeppsson, B.; Olsson, C. Mucosa-associated bacteria in two middle-aged women diagnosed with collagenous colitis. World J. Gastroenterol. 2012, 18, 1628–1634. [Google Scholar] [CrossRef]
- Helal, T.E.; Ahmed, N.S.; el Fotoh, O.A. Lymphocytic colitis: A clue to bacterial etiology. World J. Gastroenterol. 2005, 11, 7266–7271. [Google Scholar]
- Rubio, C.A.; Hubbard, G.B. Chronic colitis in baboons: Similarities with chronic colitis in humans. In Vivo 2001, 15, 109–116. [Google Scholar]
- Khalil, N.A.; Walton, G.E.; Gibson, G.R.; Tuohy, K.M.; Andrews, S.C. In vitro batch cultures of gut microbiota from healthy and ulcerative colitis (UC) subjects suggest that sulphate-reducing bacteria levels are raised in UC and by a protein-rich diet. Int. J. Food Sci. Nutr. 2014, in press. [Google Scholar]
- Kumari, R.; Ahuja, V.; Paul, J. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. World J. Gastroenterol. 2013, 19, 3404–3408. [Google Scholar] [CrossRef]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejón, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–156. [Google Scholar] [CrossRef]
- Pilarczyk-Zurek, M.; Chmielarczyk, A.; Gosiewski, T.; Tomusiak, A.; Adamski, P.; Zwolinska-Wcislo, M.; Mach, T.; Heczko, P.; Strus, M. Possible role of Escherichia coli in propagation and perpetuation of chronic inflammation in ulcerative colitis. BMC Gastroenterol. 2013. [Google Scholar] [CrossRef]
- Yukawa, T.; Ohkusa, T.; Shibuya, T.; Tsukinaga, S.; Mitobe, J.; Takakura, K.; Takahara, A.; Odahara, S.; Tajiri, H. Nested culture method improves detection of Fusobacterium from stool in patients with ulcerative colitis. Jpn. J. Infect. Dis. 2013, 66, 109–111. [Google Scholar] [CrossRef]
- Kabeerdoss, J.; Sankaran, V.; Pugazhendhi, S.; Ramakrishna, B.S. Clostridium leptum group bacteria abundance and diversity in the fecal microbiota of patients with inflammatory bowel disease: A case-control study in India. BMC Gastroenterol. 2013, 13, 20–26. [Google Scholar] [CrossRef]
- Gałecka, M.; Szachta, P.; Bartnicka, A.; Łykowska-Szuber, L.; Eder, P.; Schwiertz, A. Faecalibacterium prausnitzii and Crohn’s disease—Is there any connection? Pol. J. Microbiol. 2013, 62, 91–95. [Google Scholar]
- Kale-Pradhan, P.B.; Zhao, J.; Palmer, J.; Wilhelm, S.M. The role of antimicrobials in Crohn’s disease. Expert. Rev. Gastroenterol. Hepatol. 2013, 7, 281–288. [Google Scholar] [CrossRef]
- Nickerson, K.; McDonald, C. Crohn’s disease-associated adherent-invasive Escherichia coli adhesion is enhanced by exposure to the ubiquitous dietary polysaccharide maltodextrin. PLoS One 2012, 7, e52132. [Google Scholar] [CrossRef]
- Erickson, A.; Cantarel, B.; Lamendella, R.; Darzi, Y.; Mongodin, E.; Pan, C.; Shah, M.; Halfvarson, J.; Jansson, J.K. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One 2012, 7, e49138. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.; Sharma, Y.; Anderson, C.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Shanahan, F. The microbiota in inflammatory bowel disease: Friend, bystander, and sometime-villain. Nutr. Rev. 2012, 70, S31–S37. [Google Scholar] [CrossRef]
- Muriel-Galet, V.; Talbert, J.N.; Hernandez-Munoz, P.; Gavara, R.; Goddard, J.M. Covalent immobilization of lysozyme on ethylene vinyl alcohol films for nonmigrating antimicrobial packaging applications. J. Agric. Food Chem. 2013, 61, 6720–6727. [Google Scholar] [CrossRef]
- Ibrahim, H.R.; Imazato, K.; Ono, H. Human lysozyme possesses novel antimicrobial peptides within its N-terminal domain that target bacterial respiration. J. Agric. Food Chem. 2011, 59, 10336–10345. [Google Scholar] [CrossRef]
- Oliver, W.T.; Wells, J.E. Lysozyme as an alternative to antibiotics improves growth performance and small intestinal morphology in nursery pigs. J. Anim. Sci. 2013, 91, 3129–3136. [Google Scholar] [CrossRef]
- Teneback, C.; Scanlon, T.; Wargo, M.; Bement, J.; Griswold, K.; Leclair, L.W. Bioengineered lysozyme reduces bacterial burden and inflammation in a murine model of mucoid Pseudomonas aeruginosa lung infection. Antimicrob. Agents Chemother. 2013, 57, 5559–5564. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Klesius, P.H.; Dominowski, P.J.; Yancey, R.J.; Kievit, M.S. Chicken-type lysozyme in channel catfish: Expression analysis, lysozyme activity, and efficacy as immunostimulant against Aeromonas hydrophila infection. Fish Shellfish Immunol. 2013, 35, 680–688. [Google Scholar] [CrossRef]
- Cegielska-Radziejewska, R.; Szablewski, T. Effect of modified lysozyme on the microflora and sensory attributes of ground pork. J. Food Prot. 2013, 76, 338–342. [Google Scholar] [CrossRef]
- Lamppa, J.W.; Tanyos, S.A.; Griswold, K.E. Engineering Escherichia coli for soluble expression and single step purification of active human lysozyme. J. Biotechnol. 2013, 164, 1–8. [Google Scholar] [CrossRef]
- Rosu, V.; Bandino, E.; Cossu, A. Unraveling the transcriptional regulatory networks associated to mycobacterial cell wall defective form induction by glycine and lysozyme treatment. Microbiol. Res. 2013, 168, 153–164. [Google Scholar] [CrossRef]
- Scanlon, T.C.; Teneback, C.C.; Gill, A.; Bement, J.L.; Weiner, J.A.; Lamppa, J.; Leclair, L.W.; Griswold, K.E. Enhanced antimicrobial activity of engineered human lysozyme. ACS Chem. Biol. 2010, 5, 809–818. [Google Scholar] [CrossRef]
- Bhavsar, T.; Liu, M.; Hardej, D.; Liu, X.; Cantor, J. Aerosolized recombinant human lysozyme ameliorates Pseudomonas aeruginosa-induced pneumonia in hamsters. ACS Chem. Biol. 2010, 5, 809–818. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rubio, C.A. The Natural Antimicrobial Enzyme Lysozyme is Up-Regulated in Gastrointestinal Inflammatory Conditions. Pathogens 2014, 3, 73-92. https://doi.org/10.3390/pathogens3010073
Rubio CA. The Natural Antimicrobial Enzyme Lysozyme is Up-Regulated in Gastrointestinal Inflammatory Conditions. Pathogens. 2014; 3(1):73-92. https://doi.org/10.3390/pathogens3010073
Chicago/Turabian StyleRubio, Carlos A. 2014. "The Natural Antimicrobial Enzyme Lysozyme is Up-Regulated in Gastrointestinal Inflammatory Conditions" Pathogens 3, no. 1: 73-92. https://doi.org/10.3390/pathogens3010073
APA StyleRubio, C. A. (2014). The Natural Antimicrobial Enzyme Lysozyme is Up-Regulated in Gastrointestinal Inflammatory Conditions. Pathogens, 3(1), 73-92. https://doi.org/10.3390/pathogens3010073