
Distinct Immunological Landscapes of HCMV-Specific T Cells in Bone Marrow and Peripheral Blood




Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Samples and Donor Characteristics
2.2. Preparation of Frozen Peripheral Blood and Bone Marrow Mononuclear Cells
2.3. HCMV ORF Peptide Libraries
2.4. Major Lymphocyte Subsets Phenotyping Comparison of PBMC with BMMNC
2.5. Triple Fluorospot Assay
2.6. CMV Activation Assay and Spectral Flow Cytometry Acquisition
2.7. Analysis of Spectral Flow Data
2.7.1. Resting (Ex Vivo) T Cell Phenotype Workflow for Comparison Between PBMC and BMMNC
2.7.2. Identification of CMV Specific T Cell Responses
2.7.3. CMV IE and US28 Specific T Cell Analysis Workflow
2.8. Statistical Analysis
3. Results
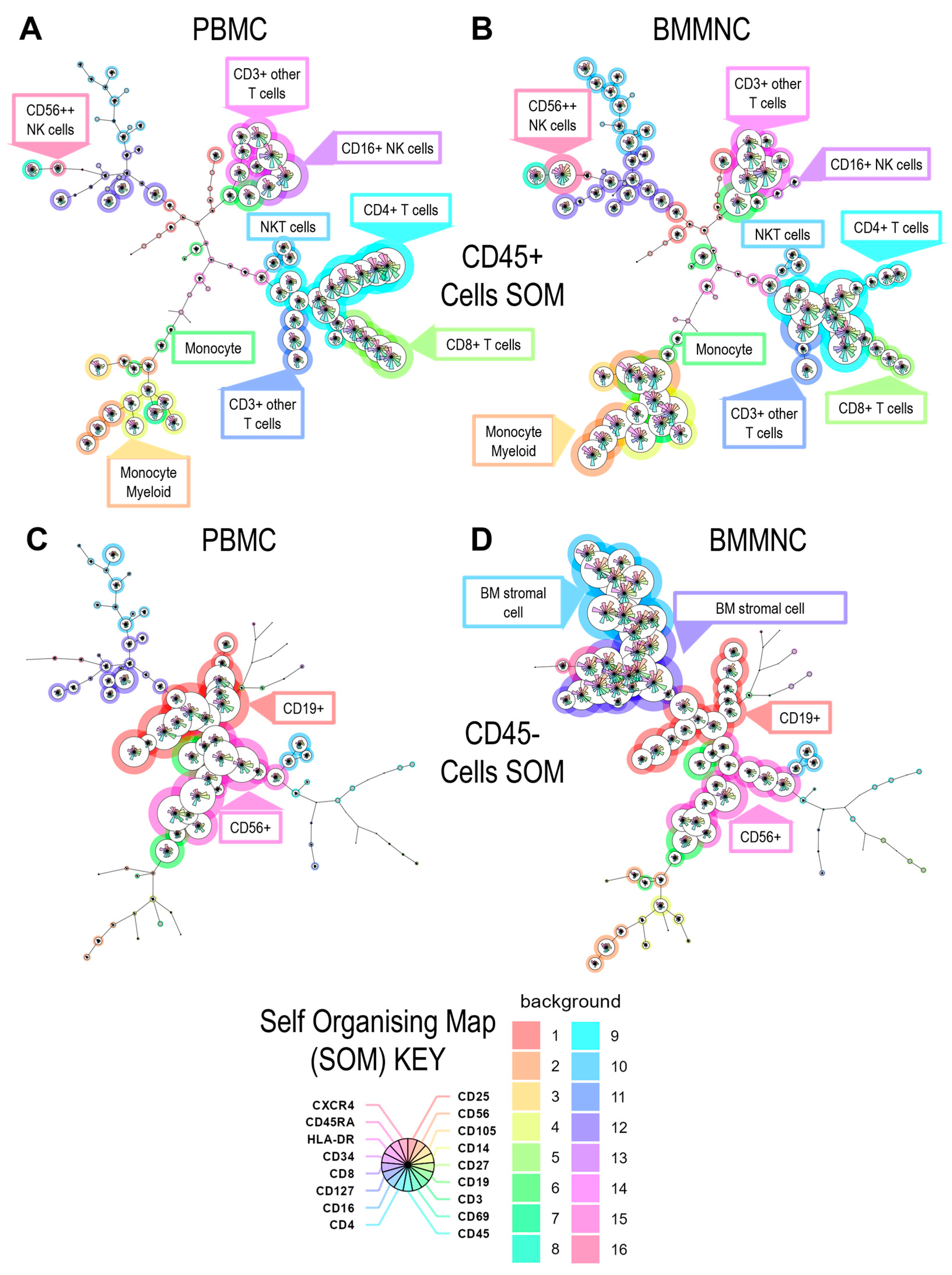
3.1. Analysis of the Cellular CD45+ and CD45− Cell Populations Present in PBMC and Bone Marrow Mononuclear Cells
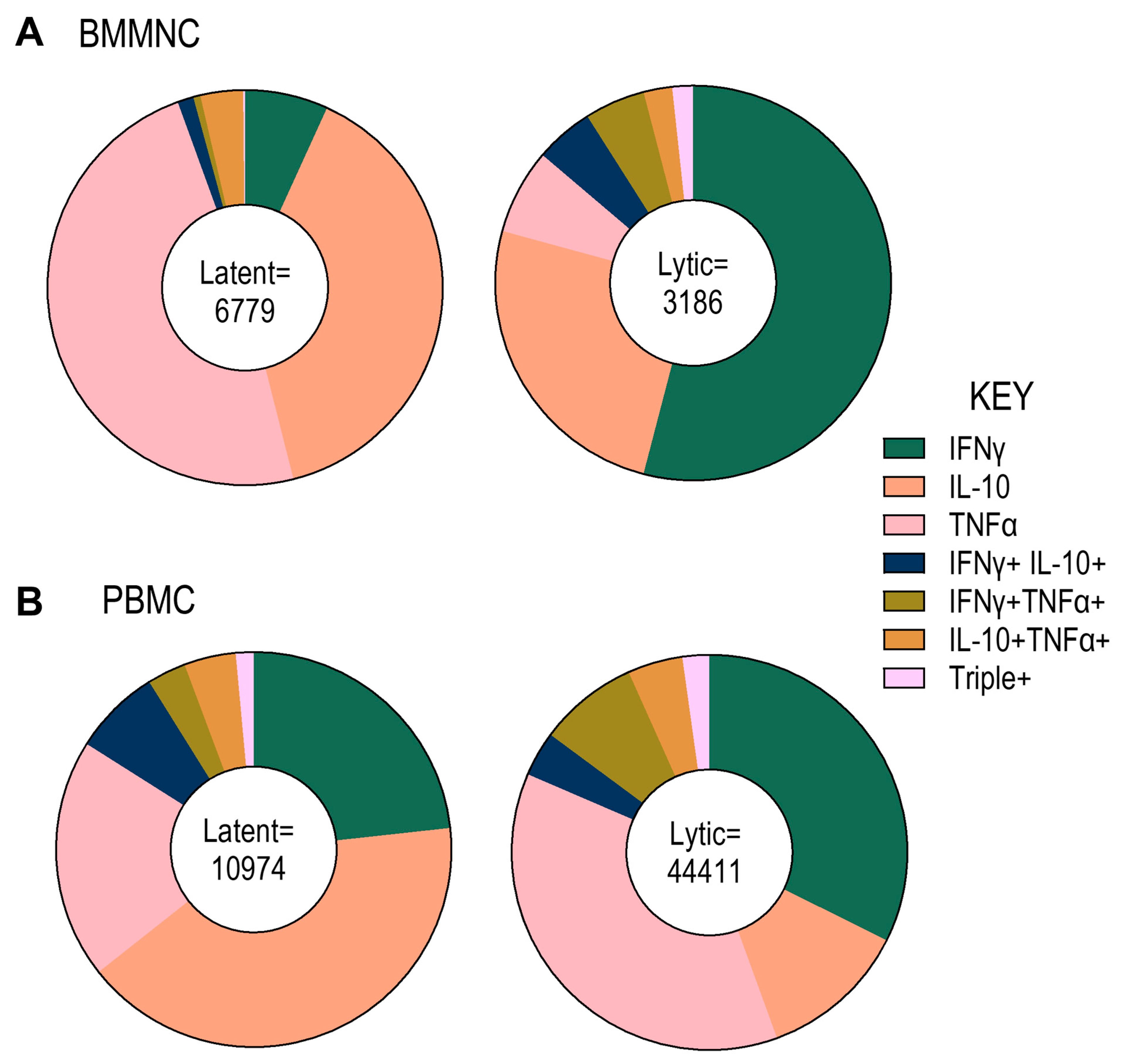
3.2. Analysis of HCMV-Specific T Cell Frequency and Cytokine Responses to Overlapping Peptide Pools Representing HCMV Lytic and Latent Proteins
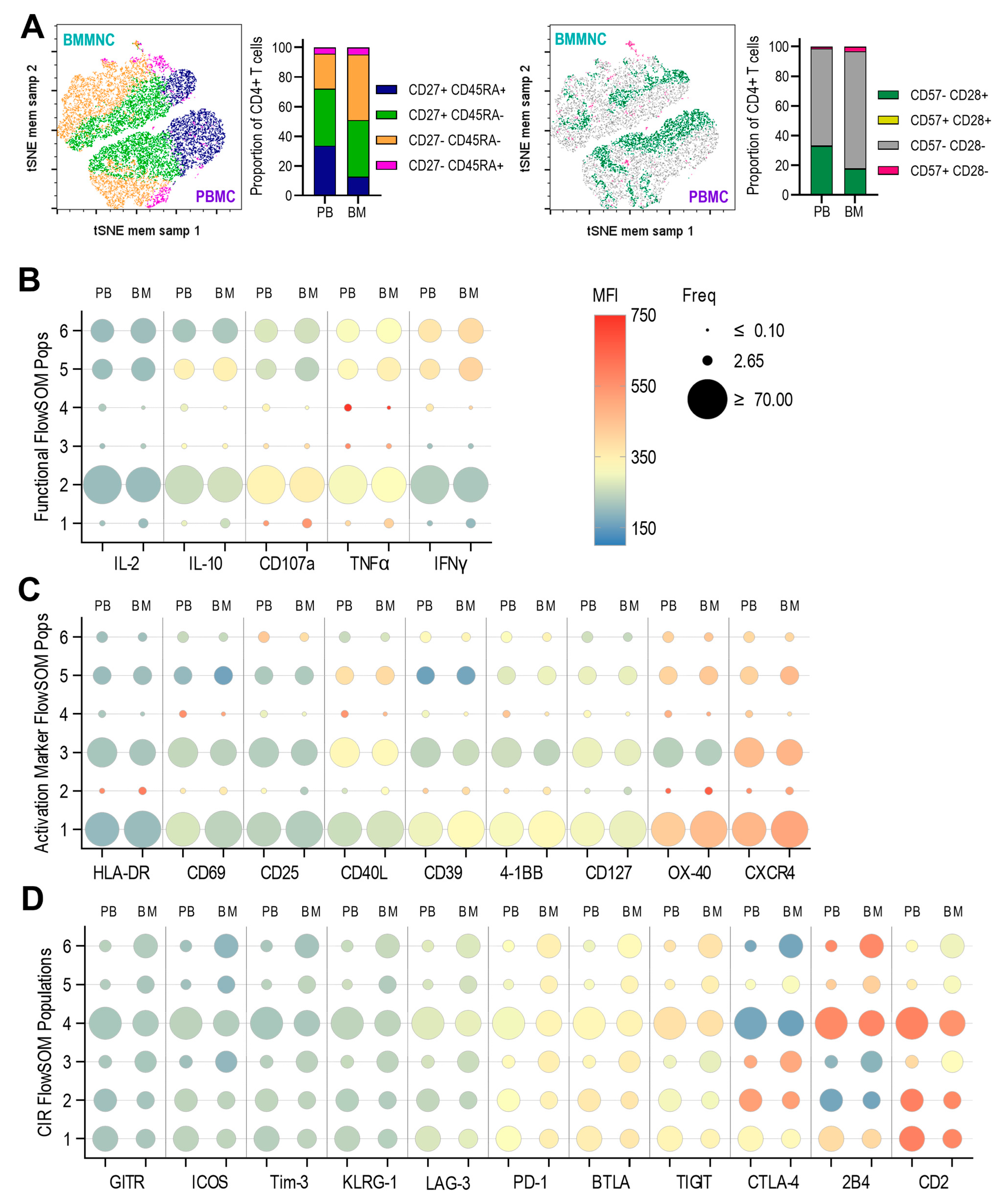
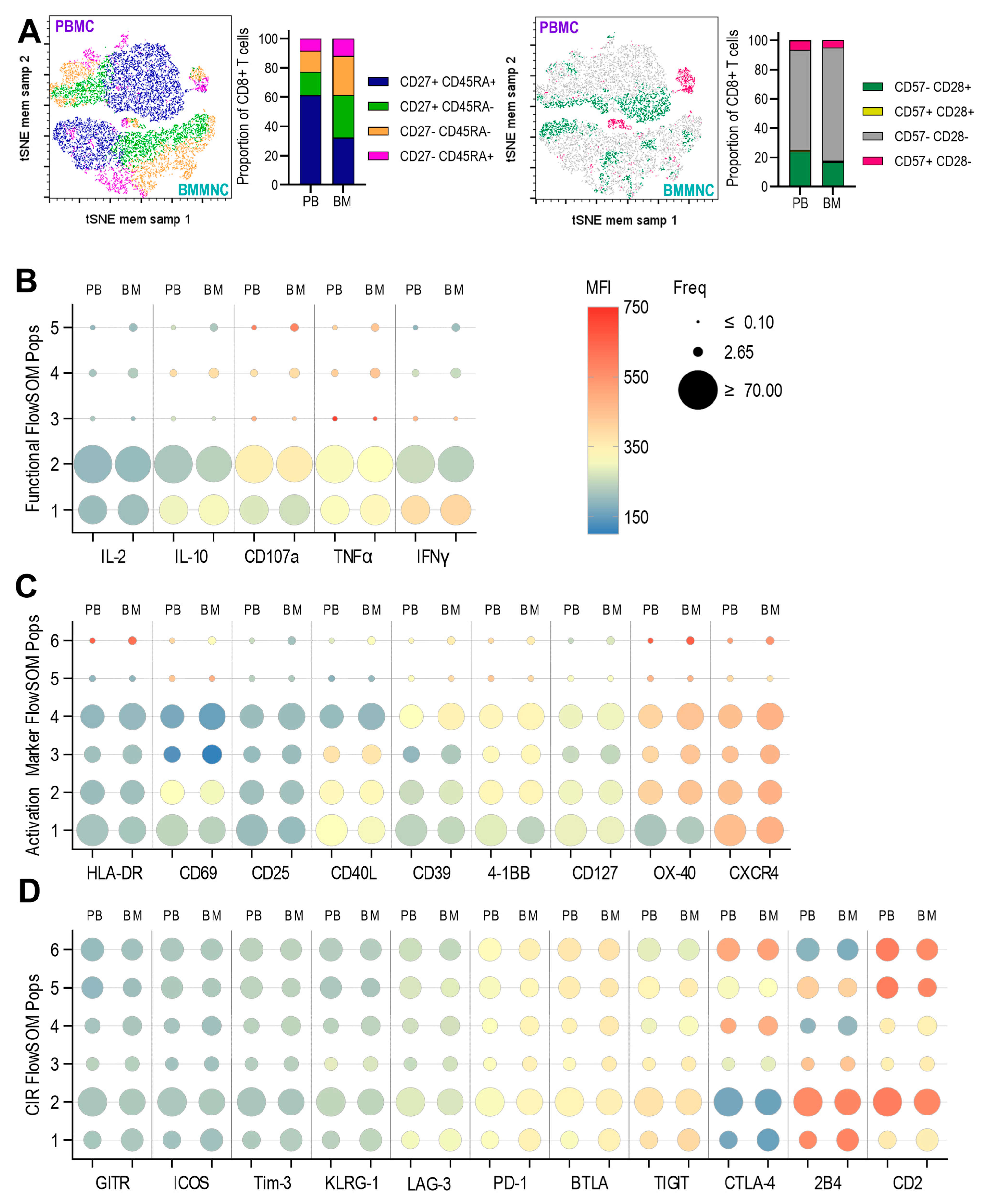
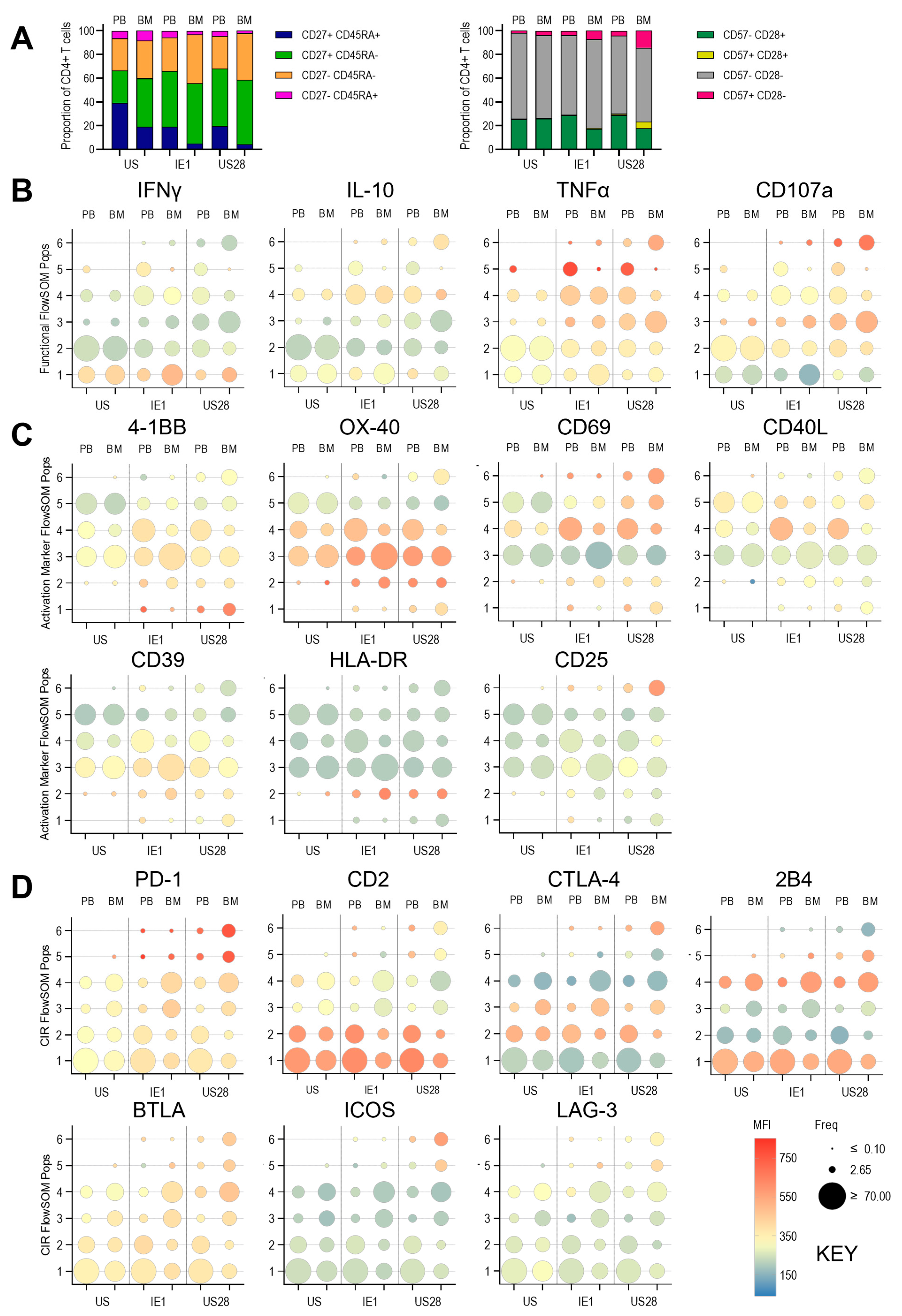
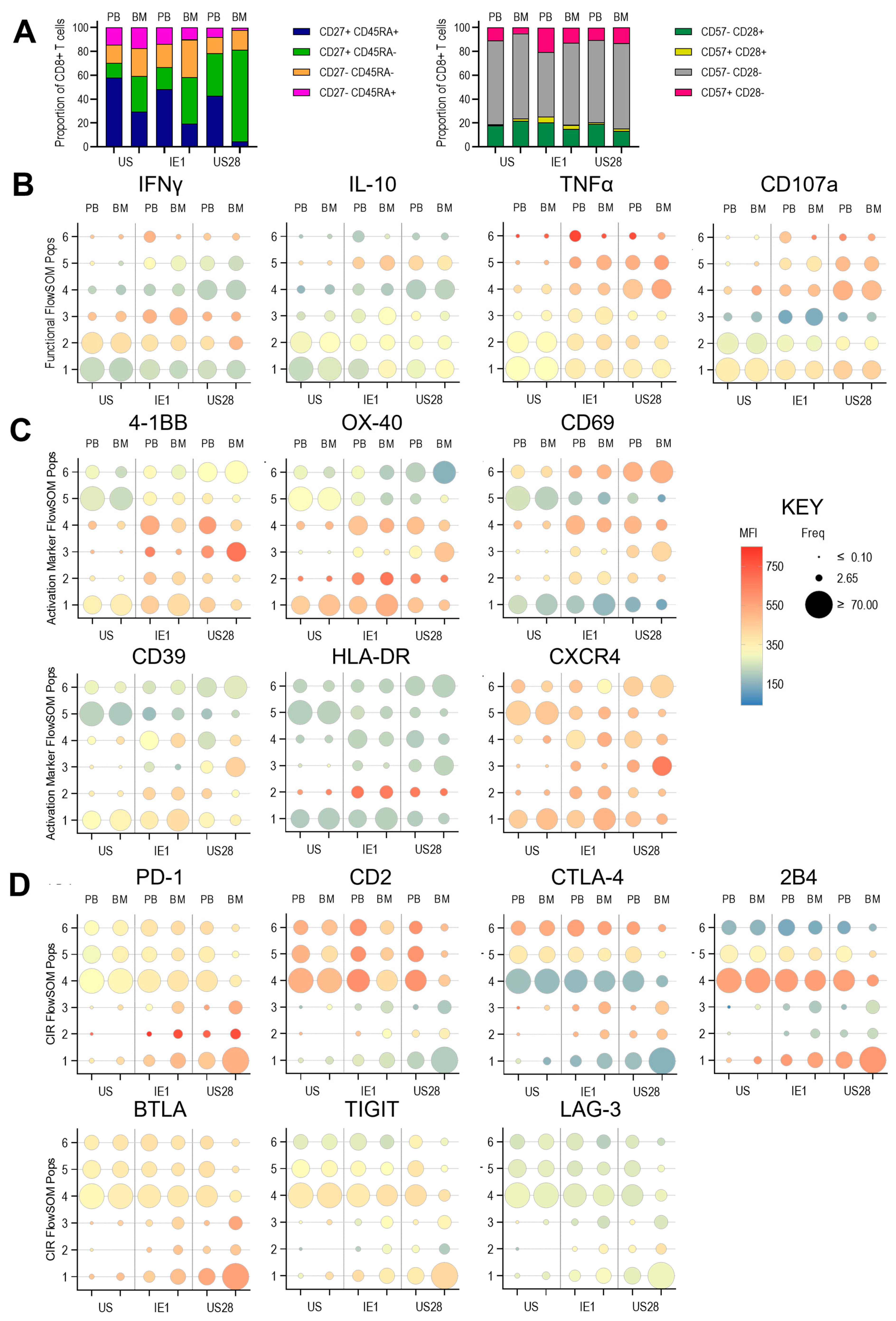
3.3. Expression of Anti-Viral Cytokines, Activation Markers, Activating and Inhibitory Checkpoint Receptors by T Cells Derived from Paired Pbmc and Bmmncs Directly Ex Vivo
3.4. Expression of Anti-Viral Cytokines, Activation Markers, Activating and Inhibitory Checkpoint Receptors in Pbmc and Bmmncs Following Hcmv Antigen Stimulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen Presentation Cells |
| BM | Bone Marrow |
| BMMNC | Bone Marrow Mononuclear Cells |
| BTLA | B and T lymphocyte attenuator |
| CIR | Checkpoint Inhibitory Receptors |
| CMV | Cytomegalovirus |
| GPCR | G protein-coupled receptor |
| HCMV | Human Cytomegalovirus |
| HDACi | Histone deacetylase inhibitors |
| HVEM | Herpesvirus entry mediator |
| IBET | bromodomain and extra-terminal domain inhibitors |
| ICOS | Inducible Costimulator |
| ITIM | Immunoreceptor Tyrosine-based Inhibitory Motif |
| iTreg | Inducible T regulatory Cell |
| LAG-3 | Lymphocyte Activation Gene 3 |
| MCMV | Murine Cytomegalovirus |
| MMR | Measles Mumps and Rubella Vaccine |
| ORFs | Open Reading Frames |
| PB | Peripheral Blood |
| PBMCs | Peripheral Blood Mononuclear cells |
| SOM | Self Organising Maps |
| TCM | T central memory cells |
| TEM | T effector memory cells |
| TEMRA | T effector memory cells re-expressing CD45RA |
| Tfh | T follicular helper cells |
| TIGIT | T cell immunoglobulin and ITIM domain |
| Tregs | T regulatory Cells |
| tSNE | t-distributed stochastic neighbour embedding |
| WLSM | Weighted Least Squares Method |
References
- Picarda, G.; Benedict, C.A. Cytomegalovirus: Shape-Shifting the Immune System. J. Immunol. 2018, 200, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Noor, M.; Lim, E.Y.; Wills, M.R. The Immune Response to Human Cytomegalovirus: Impact of Age, Co-morbidities, 3 and the Significance of Anti-viral Activity Assessment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2025. [Google Scholar] [CrossRef]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways To Maintain Latent Infection. MBio 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Elder, E.G.; Krishna, B.A.; Williamson, J.; Lim, E.Y.; Poole, E.; Sedikides, G.X.; Wills, M.; O’Connor, C.M.; Lehner, P.J.; Sinclair, J. Interferon-Responsive Genes Are Targeted during the Establishment of Human Cytomegalovirus Latency. MBio 2019, 10, e02574-19. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.L.; Kew, V.G.; Lau, J.C.H.; Murray, M.J.; Stamminger, T.; Sinclair, J.H.; Reeves, M.B. A Virally Encoded DeSUMOylase Activity Is Required for Cytomegalovirus Reactivation from Latency. Cell Rep. 2018, 24, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013, 87, 4261–4271. [Google Scholar] [CrossRef] [PubMed]
- Elder, E.; Sinclair, J. HCMV latency: What regulates the regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.M.; Jackson, S.E.; Okecha, G.; Poole, E.; Sissons, J.G.; Sinclair, J.; Wills, M.R. Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4+ T cells. PLoS Pathog. 2013, 9, e1003635. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Sedikides, G.X.; Romashova, V.; Okecha, G.; Remmerswaal, E.B.M.; Bemelman, F.J.; Sinclair, J.H.; Wills, M.R. IL-10-Secreting CD8(+) T Cells Specific for Human Cytomegalovirus (HCMV): Generation, Maintenance and Phenotype. Pathogens 2022, 11, 1530. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Sedikides, G.X.; Mason, G.M.; Okecha, G.; Wills, M.R. Human Cytomegalovirus (HCMV)-Specific CD4+ T Cells Are Polyfunctional and Can Respond to HCMV-Infected Dendritic Cells In Vitro. J. Virol. 2017, 91, 16. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Sedikides, G.X.; Okecha, G.; Poole, E.L.; Sinclair, J.H.; Wills, M.R. Latent Cytomegalovirus (CMV) Infection Does Not Detrimentally Alter T Cell Responses in the Healthy Old, But Increased Latent CMV Carriage Is Related to Expanded CMV-Specific T Cells. Front. Immunol. 2017, 8, 733. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J. Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J. Clin. Virol. 2008, 41, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.M.; Poole, E.; Sissons, J.G.; Wills, M.R.; Sinclair, J.H. Human cytomegalovirus latency alters the cellular secretome, inducing cluster of differentiation (CD)4+ T-cell migration and suppression of effector function. Proc. Natl. Acad. Sci. USA 2012, 109, 14538–14543. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Redeker, A.; Arens, R.; van Baarle, D.; van den Berg, S.P.H.; Benedict, C.A.; Cicin-Sain, L.; Hill, A.B.; Wills, M.R. CMV immune evasion and manipulation of the immune system with aging. Geroscience 2017, 39, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Poole, E.; Lau, B.; Krishna, B.; Sinclair, J.H. The immunology of human cytomegalovirus latency: Could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol. Immunol. 2015, 12, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Minton, E.J.; Tysoe, C.; Sinclair, J.H.; Sissons, J.G. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J. Virol. 1994, 68, 4017–4021. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Chen, K.C.; Groves, I.J.; Sedikides, G.X.; Gandhi, A.; Houldcroft, C.J.; Poole, E.L.; Montanuy, I.; Mason, G.M.; Okecha, G.; et al. Latent Cytomegalovirus-Driven Recruitment of Activated CD4+ T Cells Promotes Virus Reactivation. Front. Immunol. 2021, 12, 657945. [Google Scholar] [CrossRef] [PubMed]
- Okhrimenko, A.; Grun, J.R.; Westendorf, K.; Fang, Z.; Reinke, S.; von Roth, P.; Wassilew, G.; Kuhl, A.A.; Kudernatsch, R.; Demski, S.; et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc. Natl. Acad. Sci. USA 2014, 111, 9229–9234. [Google Scholar] [CrossRef] [PubMed]
- Cendon, C.; Du, W.; Durek, P.; Liu, Y.C.; Alexander, T.; Serene, L.; Yang, X.; Gasparoni, G.; Salhab, A.; Nordstrom, K.; et al. Resident memory CD4+ T lymphocytes mobilize from bone marrow to contribute to a systemic secondary immune reaction. Eur. J. Immunol. 2022, 52, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, F.; Gebhardt, T. Bone Marrow T Cells and the Integrated Functions of Recirculating and Tissue-Resident Memory T Cells. Front. Immunol. 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Dong, J. Human bone marrow-resident and blood-circulating memory T lymphocytes. Z. Rheumatol. 2018, 77, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.D.; Radbruch, A. Maintenance of quiescent immune memory in the bone marrow. Eur. J. Immunol. 2021, 51, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Hanazawa, A.; Lohning, M.; Radbruch, A.; Tokoyoda, K. CD49b/CD69-Dependent Generation of Resting T Helper Cell Memory. Front. Immunol. 2013, 4, 183. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Hauser, A.E.; Nakayama, T.; Radbruch, A. Organization of immunological memory by bone marrow stroma. Nat. Rev. Immunol. 2010, 10, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Mazo, I.B.; Honczarenko, M.; Leung, H.; Cavanagh, L.L.; Bonasio, R.; Weninger, W.; Engelke, K.; Xia, L.J.; McEver, R.P.; Koni, P.A.; et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 2005, 22, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, F.; McGrath, M.A.; Maschmeyer, P.; Bardua, M.; Lehmann, K.; Heinz, G.; Durek, P.; Heinrich, F.F.; Mashreghi, M.F.; Chang, H.D.; et al. Nonfollicular reactivation of bone marrow resident memory CD4 T cells in immune clusters of the bone marrow. Proc. Natl. Acad. Sci. USA 2018, 115, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Miron, M.; Farber, D.L. Location, location, location: Tissue resident memory T cells in mice and humans. Sci. Immunol. 2019, 4, eaas9673. [Google Scholar] [CrossRef] [PubMed]
- Pulvirenti, N.; Vasco, C.; Righetti, C.; Dadova, P.; Boffa, G.; Laroni, A.; Vigo, T.; Raiola, A.M.; Crosti, M.C.; Maglie, S.; et al. The Human Bone Marrow May Offer an IL-15-Dependent Survival Niche for EOMES+ Tr1-Like Cells. Eur. J. Immunol. 2025, 55, e202451644. [Google Scholar] [CrossRef] [PubMed]
- Schneider Revueltas, E.; Ferreira-Gomes, M.; Guerra, G.M.; Durek, P.; Heinrich, F.; Casanovas Subirana, A.; Tokoyoda, K.; Dong, J.; Reinke, S.; Hardt, S.; et al. Surface CD69-Negative CD4 and CD8 Bone Marrow-Resident Human Memory T Cells. Eur. J. Immunol. 2025, 55, e202451529. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Sedikides, G.X.; Okecha, G.; Wills, M.R. Generation, maintenance and tissue distribution of T cell responses to human cytomegalovirus in lytic and latent infection. Med. Microbiol. Immunol. 2019, 208, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.L.; Miron, M.; Thome, J.J.; Matsuoka, N.; Weiner, J.; Rak, M.A.; Igarashi, S.; Granot, T.; Lerner, H.; Goodrum, F.; et al. Tissue reservoirs of antiviral T cell immunity in persistent human CMV infection. J. Exp. Med. 2017, 214, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Palendira, U.; Chinn, R.; Raza, W.; Piper, K.; Pratt, G.; Machado, L.; Bell, A.; Khan, N.; Hislop, A.D.; Steyn, R.; et al. Selective accumulation of virus-specific CD8(+) T cells with unique homing phenotype within the human bone marrow. Blood 2008, 112, 3293–3302. [Google Scholar] [CrossRef] [PubMed]
- Letsch, A.; Knoedler, M.; Na, I.K.; Kern, F.; Asemissen, A.M.; Keilholz, U.; Loesch, M.; Thiel, E.; Volk, H.D.; Scheibenbogen, C. CMV-specific central memory T cells reside in bone marrow. Eur. J. Immunol. 2007, 37, 3063–3068. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Geerman, S.; Collins, N.; Brasser, G.; Nota, B.; Stark, R.; Behr, F.; Oja, A.; Slot, E.; Panagioti, E.; et al. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8+ T cells in the bone marrow. Eur. J. Immunol. 2019, 49, 853–872. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Mason, G.M.; Okecha, G.; Sissons, J.G.; Wills, M.R. Diverse specificities, phenotypes, and antiviral activities of cytomegalovirus-specific CD8+ T cells. J. Virol. 2014, 88, 10894–10908. [Google Scholar] [CrossRef] [PubMed]
- Roca, C.P.; Burton, O.T.; Gergelits, V.; Prezzemolo, T.; Whyte, C.E.; Halpert, R.; Kreft, L.; Collier, J.; Botzki, A.; Spidlen, J.; et al. AutoSpill is a principled framework that simplifies the analysis of multichromatic flow cytometry data. Nat. Commun. 2021, 12, 2890. [Google Scholar] [CrossRef] [PubMed]
- Emmaneel, A.; Quintelier, K.; Sichien, D.; Rybakowska, P.; Maranon, C.; Alarcon-Riquelme, M.E.; Van Isterdael, G.; Van Gassen, S.; Saeys, Y. PeacoQC: Peak-based selection of high quality cytometry data. Cytom. Part A 2022, 101, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.C.; Ciccolella, C.O.; Anno, R.; Halpert, R.; Spidlen, J.; Snyder-Cappione, J.E. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 2019, 10, 5415. [Google Scholar] [CrossRef] [PubMed]
- Linderman, G.C.; Rachh, M.; Hoskins, J.G.; Steinerberger, S.; Kluger, Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 2019, 16, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Tippalagama, R.; Chihab, L.Y.; Kearns, K.; Lewis, S.; Panda, S.; Willemsen, L.; Burel, J.G.; Lindestam Arlehamn, C.S. Antigen-specificity measurements are the key to understanding T cell responses. Front. Immunol. 2023, 14, 1127470. [Google Scholar] [CrossRef] [PubMed]
- Poloni, C.; Schonhofer, C.; Ivison, S.; Levings, M.K.; Steiner, T.S.; Cook, L. T-cell activation-induced marker assays in health and disease. Immunol. Cell Biol. 2023, 101, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Altosole, T.; Rotta, G.; Uras, C.R.M.; Bornheimer, S.J.; Fenoglio, D. An optimized flow cytometry protocol for simultaneous detection of T cell activation induced markers and intracellular cytokines: Application to SARS-CoV-2 immune individuals. J. Immunol. Methods 2023, 515, 113443. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, A.; Sannier, G.; Nicolas, A.; Nayrac, M.; Delgado, G.G.; Cloutier, R.; Brassard, N.; Laporte, M.; Duchesne, M.; Sreng Flores, A.M.; et al. Enhanced detection of antigen-specific T cells by a multiplexed AIM assay. Cell Rep. Methods 2024, 4, 100690. [Google Scholar] [CrossRef] [PubMed]
- Porwit, A.; Béné, M.-C. Flow Cytometry of Normal Blood, Bone Marrow and Lymphatic Tissue. In Multiparameter Flow Cytometry in the Diagnosis of Hematologic Malignancies; Porwit, A., Béné, M.C., Eds.; Cambridge University Press: Cambridge, UK, 2018; pp. 36–60. [Google Scholar]
- Carvalho, J.M.; Souza, M.K.; Buccheri, V.; Rubens, C.V.; Kerbauy, J.; Oliveira, J.S. CD34-positive cells and their subpopulations characterized by flow cytometry analyses on the bone marrow of healthy allogenic donors. Sao Paulo Med. J. 2009, 127, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.C.; Souvignet, A.; Pont, J.; Solly, F.; Mondet, J.; Kesr, S.; Pernollet, M.; Dumestre-Perard, C.; Campos, L.; Cesbron, J.Y. One tube with eight antibodies for 14-part bone marrow leukocyte differential using flow cytometry. Cytom. Part B Clin. Cytom. 2017, 92, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Duffy, M.P.; Ahn, K.J.; Sussman, J.H.; Pang, M.X.; Smith, D.; Duncan, G.; Zhang, I.R.; Huang, J.F.; Lin, Y.L.; et al. Mapping the cellular biogeography of human bone marrow niches using single-cell transcriptomics and proteomic imaging. Cell 2024, 187, 3120–3140.e29. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Q.; Shi, J.; Xu, X.; Xu, J. The role of TNF-alpha in the fate regulation and functional reprogramming of mesenchymal stem cells in an inflammatory microenvironment. Front. Immunol. 2023, 14, 1074863. [Google Scholar]
- Mancusi, A.; Piccinelli, S.; Velardi, A.; Pierini, A. The Effect of TNF-alpha on Regulatory T Cell Function in Graft-versus-Host Disease. Front. Immunol. 2018, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Kuo, C.T.; Leiden, J.M. Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nat. Immunol. 2001, 2, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Zhang, Z.; Thorp, E.B.; Hummel, M. Cytomegalovirus Latency and Reactivation: An Intricate Interplay With the Host Immune Response. Front. Cell. Infect. Microbiol. 2020, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Lau, B.; Jackson, S.E.; Wills, M.R.; Sinclair, J.H.; Poole, E. Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci. Rep. 2016, 6, 24674. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain proteins regulate human cytomegalovirus latency and reactivation allowing epigenetic therapeutic intervention. Proc. Natl. Acad. Sci. USA 2021, 118, e2023025118. [Google Scholar] [CrossRef] [PubMed]
- De Groof, T.W.M.; Elder, E.G.; Lim, E.Y.; Heukers, R.; Bergkamp, N.D.; Groves, I.J.; Wills, M.; Sinclair, J.H.; Smit, M.J. Targeting the latent human cytomegalovirus reservoir for T-cell-mediated killing with virus-specific nanobodies. Nat. Commun. 2021, 12, 4436. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Miller, W.E.; O’Connor, C.M. US28: HCMV’s Swiss Army Knife. Viruses 2018, 10, 445. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Spiess, K.; Poole, E.L.; Lau, B.; Voigt, S.; Kledal, T.N.; Rosenkilde, M.M.; Sinclair, J.H. Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat. Commun. 2017, 8, 14321. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Poole, E.; Groves, I.; Owen, D.J.; Graham, S.C.; Sinclair, J.; Kelly, B.T. Repurposing an endogenous degradation domain for antibody-mediated disposal of cell-surface proteins. EMBO Rep. 2024, 25, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, T.; Ho, G.T.; Pump, W.C.; Blasczyk, R.; Bade-Doeding, C. Battle between Host Immune Cellular Responses and HCMV Immune Evasion. Int. J. Mol. Sci. 2019, 20, 3626. [Google Scholar] [CrossRef] [PubMed]
- Noriega, V.; Redmann, V.; Gardner, T.; Tortorella, D. Diverse immune evasion strategies by human cytomegalovirus. Immunol. Res. 2012, 54, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Garcia, W.; Godwin, M.J.; Spencer, J.V.; Stern, J.L.; Abendroth, A.; Slobedman, B. Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J. Virol. 2008, 82, 3736–3750. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Ural, B.B.; Farber, D.L. Tissue-specific immunity for a changing world. Cell 2021, 184, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.M.; Dowell, A.C.; Zuo, J.; Verma, K.; Kinsella, F.A.M.; Begum, J.; Croft, W.; Sharma-Oates, A.; Pratt, G.; Moss, P. PD-1 is imprinted on cytomegalovirus-specific CD4+ T cells and attenuates Th1 cytokine production whilst maintaining cytotoxicity. PLoS Pathog. 2021, 17, e1009349. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Snyder, C.M. Inhibitory Molecules PD-1, CD73 and CD39 Are Expressed by CD8(+) T Cells in a Tissue-Dependent Manner and Can Inhibit T Cell Responses to Stimulation. Front. Immunol. 2021, 12, 704862. [Google Scholar] [CrossRef] [PubMed]
- Theobald, S.J.; Khailaie, S.; Meyer-Hermann, M.; Volk, V.; Olbrich, H.; Danisch, S.; Gerasch, L.; Schneider, A.; Sinzger, C.; Schaudien, D.; et al. Signatures of T and B Cell Development, Functional Responses and PD-1 Upregulation After HCMV Latent Infections and Reactivations in Nod.Rag.Gamma Mice Humanized With Cord Blood CD34+ Cells. Front. Immunol. 2018, 9, 2734. [Google Scholar] [CrossRef] [PubMed]
- Dirks, J.; Tas, H.; Schmidt, T.; Kirsch, S.; Gartner, B.C.; Sester, U.; Sester, M. PD-1 analysis on CD28− CD27− CD4 T cells allows stimulation-independent assessment of CMV viremic episodes in transplant recipients. Am. J. Transplant. 2013, 13, 3132–3141. [Google Scholar] [CrossRef] [PubMed]
- Gallez-Hawkins, G.M.; Thao, L.; Palmer, J.; Dagis, A.; Li, X.; Franck, A.E.; Tegtmeier, B.; Lacey, S.F.; Diamond, D.J.; Forman, S.J.; et al. Increased programmed death-1 molecule expression in cytomegalovirus disease and acute graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2009, 15, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Tovar-Salazar, A.; Weinberg, A. Understanding the mechanism of action of cytomegalovirus-induced regulatory T cells. Virology 2020, 547, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.A.; Kronenberg, K.; Riquelme, P.; Wenzel, J.J.; Glehr, G.; Schilling, H.L.; Zeman, F.; Evert, K.; Schmiedel, M.; Mickler, M.; et al. Virus-specific memory T cell responses unmasked by immune checkpoint blockade cause hepatitis. Nat. Commun. 2021, 12, 1439. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; June, C.H. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. J. Clin. Investig. 2010, 120, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Quezada, S.A.; Allison, J.P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 2010, 22, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Liu, K.; Xiong, H. Roles of BTLA in Immunity and Immune Disorders. Front. Immunol. 2021, 12, 654960. [Google Scholar] [CrossRef] [PubMed]
- Serriari, N.E.; Gondois-Rey, F.; Guillaume, Y.; Remmerswaal, E.B.; Pastor, S.; Messal, N.; Truneh, A.; Hirsch, I.; van Lier, R.A.; Olive, D. B and T lymphocyte attenuator is highly expressed on CMV-specific T cells during infection and regulates their function. J. Immunol. 2010, 185, 3140–3148. [Google Scholar] [CrossRef] [PubMed]
- Bessa, V.; Sun, M.; Meier, M.; Zeng, Y.; Xu, S.; Dolff, S.; Sommerwerck, U.; Korth, J.; Taube, C.; Aigner, C.; et al. Expression pattern of co-inhibitory molecules on CMV-specific T-cells in lung transplant patients. Clin. Immunol. 2019, 208, 108258. [Google Scholar] [CrossRef] [PubMed]
- Dornieden, T.; Wilde, B.; Korth, J.; Werner, K.; Horn, P.A.; Witzke, O.; Lindemann, M. Enhancement of Cytomegalovirus-Specific Cytokine Production after Modulation of the Costimulation in Kidney Transplant Patients. J. Immunol. Res. 2019, 2019, 3926175. [Google Scholar] [CrossRef] [PubMed]
- Bitra, A.; Nemcovicova, I.; Picarda, G.; Doukov, T.; Wang, J.; Benedict, C.A.; Zajonc, D.M. Structure of human cytomegalovirus UL144, an HVEM orthologue, bound to the B and T cell lymphocyte attenuator. J. Biol. Chem. 2019, 294, 10519–10529. [Google Scholar] [CrossRef] [PubMed]
- Waller, E.C.; McKinney, N.; Hicks, R.; Carmichael, A.J.; Sissons, J.G.; Wills, M.R. Differential costimulation through CD137 (4–1BB) restores proliferation of human virus-specific “effector memory” (CD28− CD45RAHI) CD8+ T cells. Blood 2007, 110, 4360–4366. [Google Scholar] [CrossRef] [PubMed]
- Bruno, F.; Fornara, C.; Zelini, P.; Furione, M.; Carrara, E.; Scaramuzzi, L.; Cane, I.; Mele, F.; Sallusto, F.; Lilleri, D.; et al. Follicular helper T-cells and virus-specific antibody response in primary and reactivated human cytomegalovirus infections of the immunocompetent and immunocompromised transplant patients. J. Gen. Virol. 2016, 97, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Angulo, G.; Zeleznjak, J.; Martinez-Vicente, P.; Punet-Ortiz, J.; Hengel, H.; Messerle, M.; Oxenius, A.; Jonjic, S.; Krmpotic, A.; Engel, P.; et al. Cytomegalovirus restricts ICOSL expression on antigen-presenting cells disabling T cell co-stimulation and contributing to immune evasion. Elife 2021, 10, e59350. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Marsden, M.; Stacey, M.A.; Abdul-Karim, J.; Gimeno Brias, S.; Costa Bento, D.; Scurr, M.J.; Ghazal, P.; Weaver, C.T.; Carlesso, G.; et al. Cytomegalovirus-Specific IL-10-Producing CD4+ T Cells Are Governed by Type-I IFN-Induced IL-27 and Promote Virus Persistence. PLoS Pathog. 2016, 12, e1006050. [Google Scholar] [CrossRef] [PubMed]
- Harjunpaa, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; d’Alessandro, M.; Cavallaro, D.; Pordon, E.; Cassai, L.; Gangi, S.; Meloni, F.; Montagnani, F.; Paladini, P.; Refini, R.M.; et al. Immune checkpoint analysis of T-cell responses to pp65 and IE-1 antigens in end-stage lung diseases. Scand. J. Immunol. 2023, 97, e13248. [Google Scholar] [CrossRef] [PubMed]
- Pickering, H.; Sen, S.; Cappelletti, M.; Lum, E.L.; Bunnapradist, S.; Reed, E.F.; Schaenman, J.M. TIM3 and TIGIT-expressing CD4 T cells are impacted by kidney transplantation and associated with risk of infection. Front. Immunol. 2025, 16, 1550154. [Google Scholar] [CrossRef] [PubMed]
- Schorer, M.; Rakebrandt, N.; Lambert, K.; Hunziker, A.; Pallmer, K.; Oxenius, A.; Kipar, A.; Stertz, S.; Joller, N. TIGIT limits immune pathology during viral infections. Nat. Commun. 2020, 11, 1288. [Google Scholar] [CrossRef] [PubMed]
- Aghbash, P.S.; Rasizadeh, R.; Arefi, V.; Nahand, J.S.; Baghi, H.B. Immune-checkpoint expression in antigen-presenting cells (APCs) of cytomegaloviruses infection after transplantation: As a diagnostic biomarker. Arch. Microbiol. 2023, 205, 280. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Workman, C.J.; Vignali, D.A.A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Anderson, A.C.; Kuchroo, V.K. LAG-3, TIM-3, and TIGIT: Distinct functions in immune regulation. Immunity 2024, 57, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, A.; Qiu, C.; Wang, W.; Yang, Y.; Qiu, C.; Liu, A.; Zhu, L.; Yuan, S.; Hu, H.; et al. The upregulation of LAG-3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J. Immunol. 2015, 194, 3873–3882. [Google Scholar] [CrossRef] [PubMed]
- Rousseliere, A.; Gerard, N.; Delbos, L.; Guerif, P.; Giral, M.; Bressollette-Bodin, C.; Charreau, B. Distinctive phenotype for HLA-E- versus HLA-A2-restricted memory CD8 αβT cells in the course of HCMV infection discloses features shared with NKG2C+CD57+NK and δ2−γδT cell subsets. Front. Immunol. 2022, 13, 1063690. [Google Scholar] [CrossRef] [PubMed]
- Ogando-Rivas, E.; Castillo, P.; Jones, N.; Trivedi, V.; Drake, J.; Dechkovskaia, A.; Candelario, K.M.; Yang, C.; Mitchell, D.A. Effects of immune checkpoint blockade on antigen-specific CD8+ T cells for use in adoptive cellular therapy. Microbiol. Immunol. 2022, 66, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Timperi, E.; Barnaba, V. CD39 Regulation and Functions in T Cells. Int. J. Mol. Sci. 2021, 22, 8068. [Google Scholar] [CrossRef] [PubMed]
- Schwele, S.; Fischer, A.M.; Brestrich, G.; Wlodarski, M.W.; Wagner, L.; Schmueck, M.; Roemhild, A.; Thomas, S.; Hammer, M.H.; Babel, N.; et al. Cytomegalovirus-specific regulatory and effector T cells share TCR clonality--possible relation to repetitive CMV infections. Am. J. Transplant. 2012, 12, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.F.; Degefu, H.N.; Chen, T.; Kleist, S.A.; Musial, S.C.; Ford, M.A.; Searles, T.G.; Lin, C.C.; Skorput, A.G.J.; Shirai, K.; et al. CD39 Is Expressed on Functional Effector and Tissue-resident Memory CD8+ T Cells. J. Immunol. 2024, 213, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.L.; Noor, M.; Lim, E.Y.; Houldcroft, C.J.; Okecha, G.; Atkinson, C.; Reeves, M.B.; Jackson, S.E.; Wills, M.R. HCMV carriage in the elderly diminishes anti-viral functionality of the adaptive immune response resulting in virus replication at peripheral sites. Front. Immunol. 2022, 13, 1083230. [Google Scholar] [CrossRef] [PubMed]
- Durek, P.; Nordstrom, K.; Gasparoni, G.; Salhab, A.; Kressler, C.; de Almeida, M.; Bassler, K.; Ulas, T.; Schmidt, F.; Xiong, J.; et al. Epigenomic Profiling of Human CD4+ T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development. Immunity 2016, 45, 1148–1161. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor ID | Age | Sex | HLA Class I |
|---|---|---|---|
| 45739 | 25 | Male | A01 A32; B08 B41; C07 C17 |
| 45855 | 31 | Male | A03 A30; B35 B81; C04 C08 |
| 46899 | 24 | Male | A31 A36; B15 B53; C04 C08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, S.E.; Fairclough, R.; Romashova, V.; Okecha, G.; Wills, M.R. Distinct Immunological Landscapes of HCMV-Specific T Cells in Bone Marrow and Peripheral Blood. Pathogens 2025, 14, 722. https://doi.org/10.3390/pathogens14080722
Jackson SE, Fairclough R, Romashova V, Okecha G, Wills MR. Distinct Immunological Landscapes of HCMV-Specific T Cells in Bone Marrow and Peripheral Blood. Pathogens. 2025; 14(8):722. https://doi.org/10.3390/pathogens14080722
Chicago/Turabian StyleJackson, Sarah E., Rosie Fairclough, Veronika Romashova, Georgina Okecha, and Mark R. Wills. 2025. "Distinct Immunological Landscapes of HCMV-Specific T Cells in Bone Marrow and Peripheral Blood" Pathogens 14, no. 8: 722. https://doi.org/10.3390/pathogens14080722
APA StyleJackson, S. E., Fairclough, R., Romashova, V., Okecha, G., & Wills, M. R. (2025). Distinct Immunological Landscapes of HCMV-Specific T Cells in Bone Marrow and Peripheral Blood. Pathogens, 14(8), 722. https://doi.org/10.3390/pathogens14080722