The Microbiome Characterization of Edible Visceral Organs and Fresh Meat During Production in a Pig Processing Facility in Thailand

 ,
,  , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Animals and Sampling
2.3. Sample Collection
2.4. DNA Extraction
2.5. 16S rRNA Amplicon Sequencing and Bioinformatics
2.6. Statistics
3. Results
3.1. Bacterial Community Structure
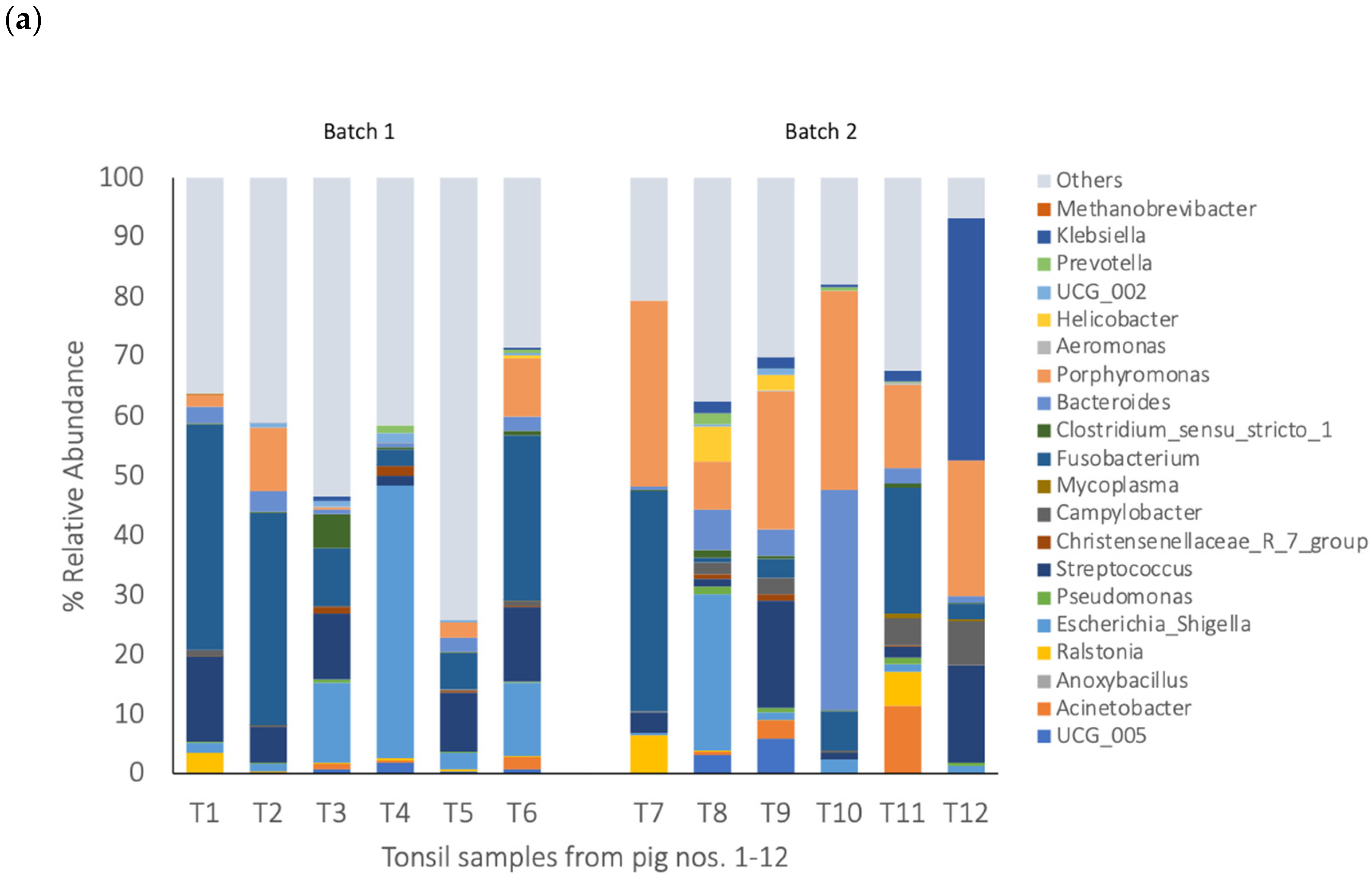
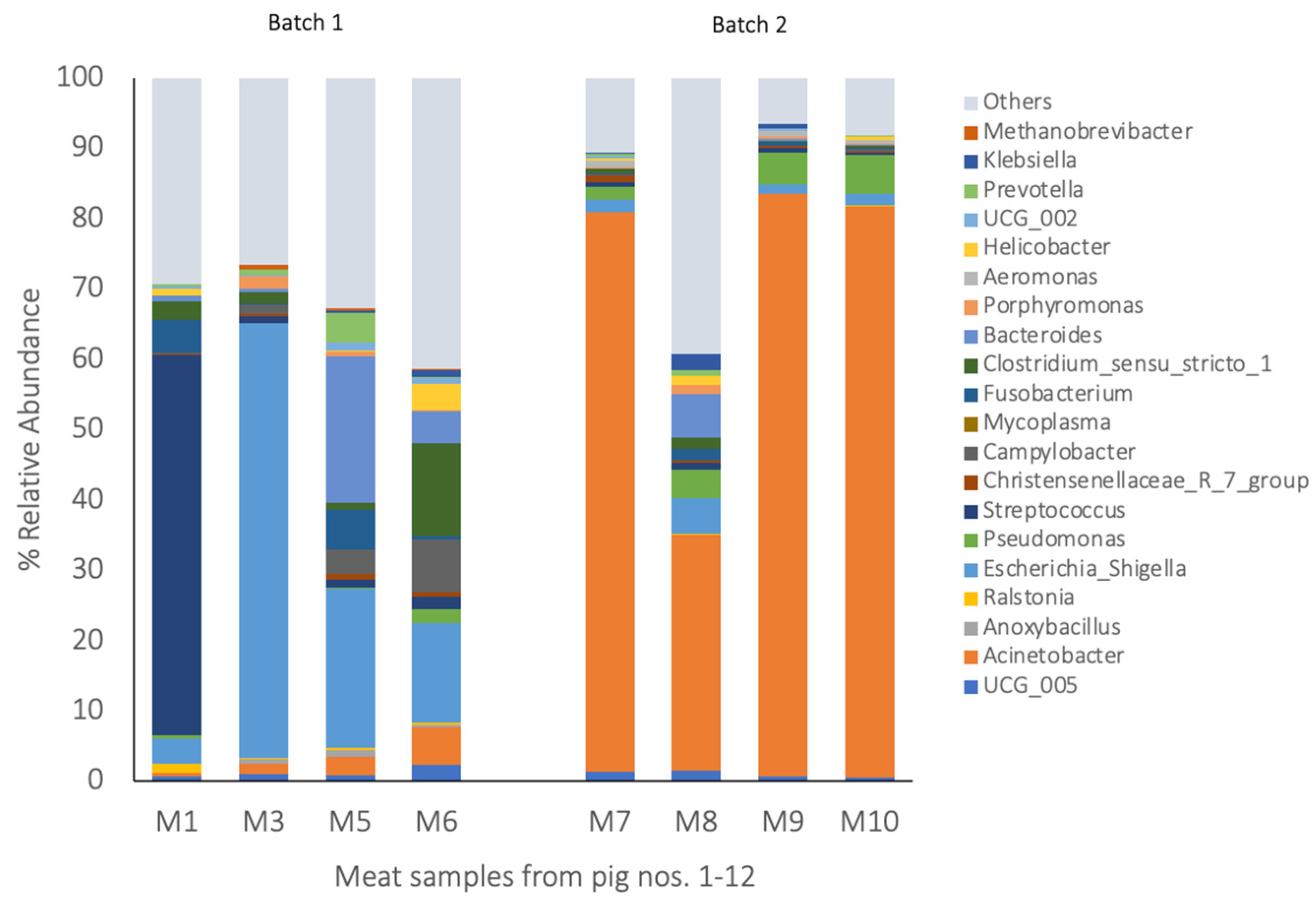
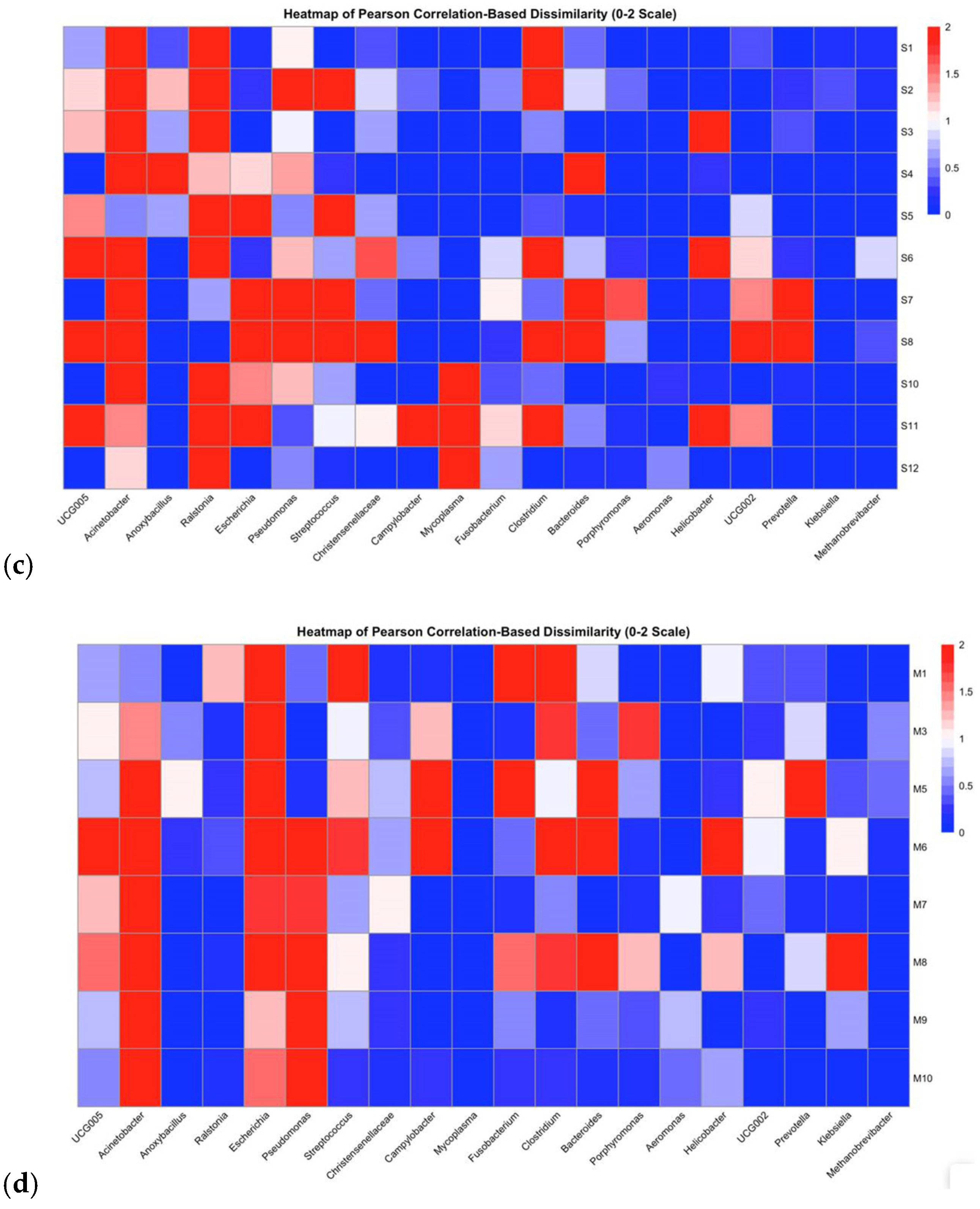
3.2. Bacterial Composition of Blood, Visceral Organs, and Meat
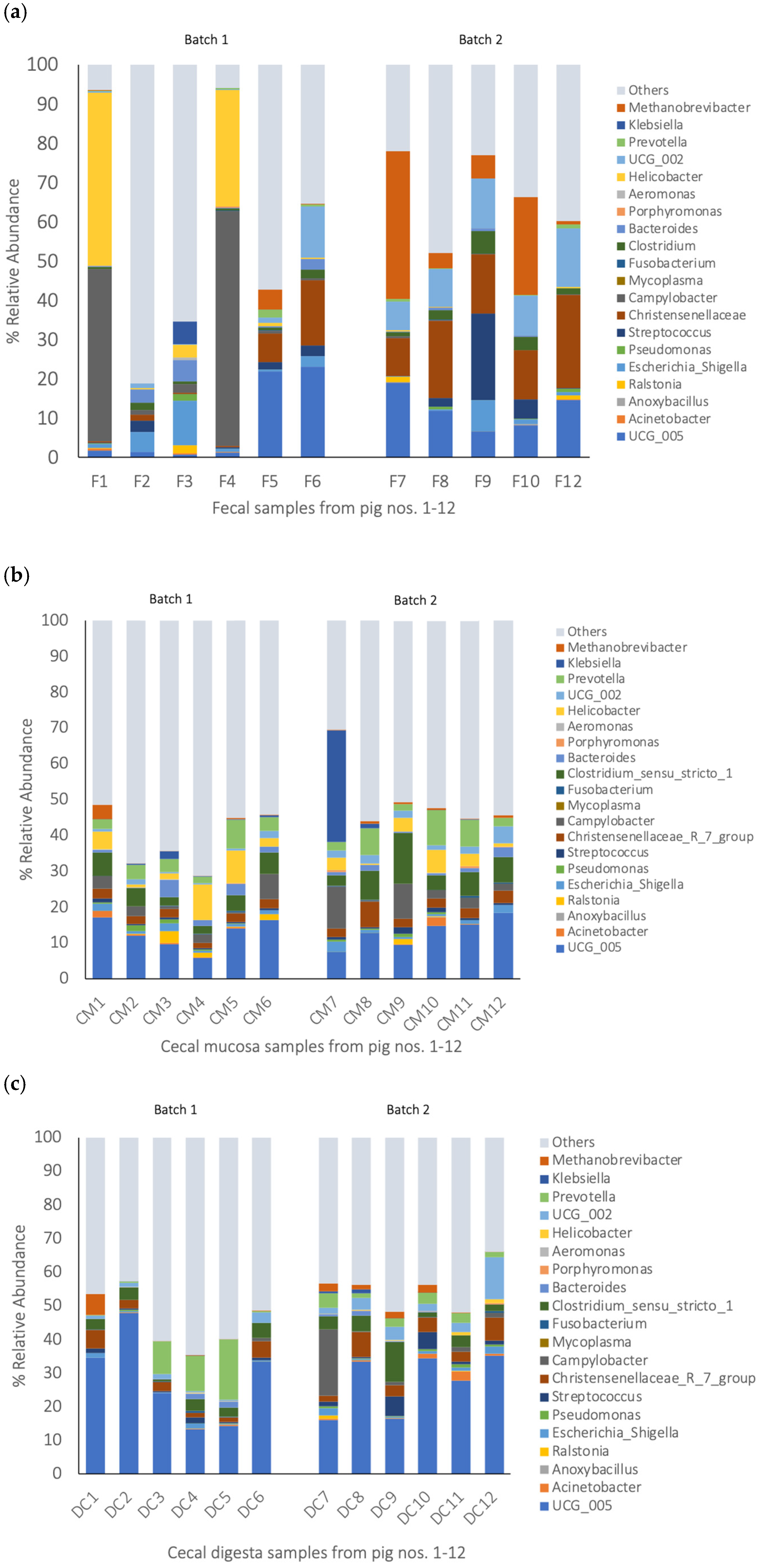
3.3. Bacterial Composition of Gut Samples
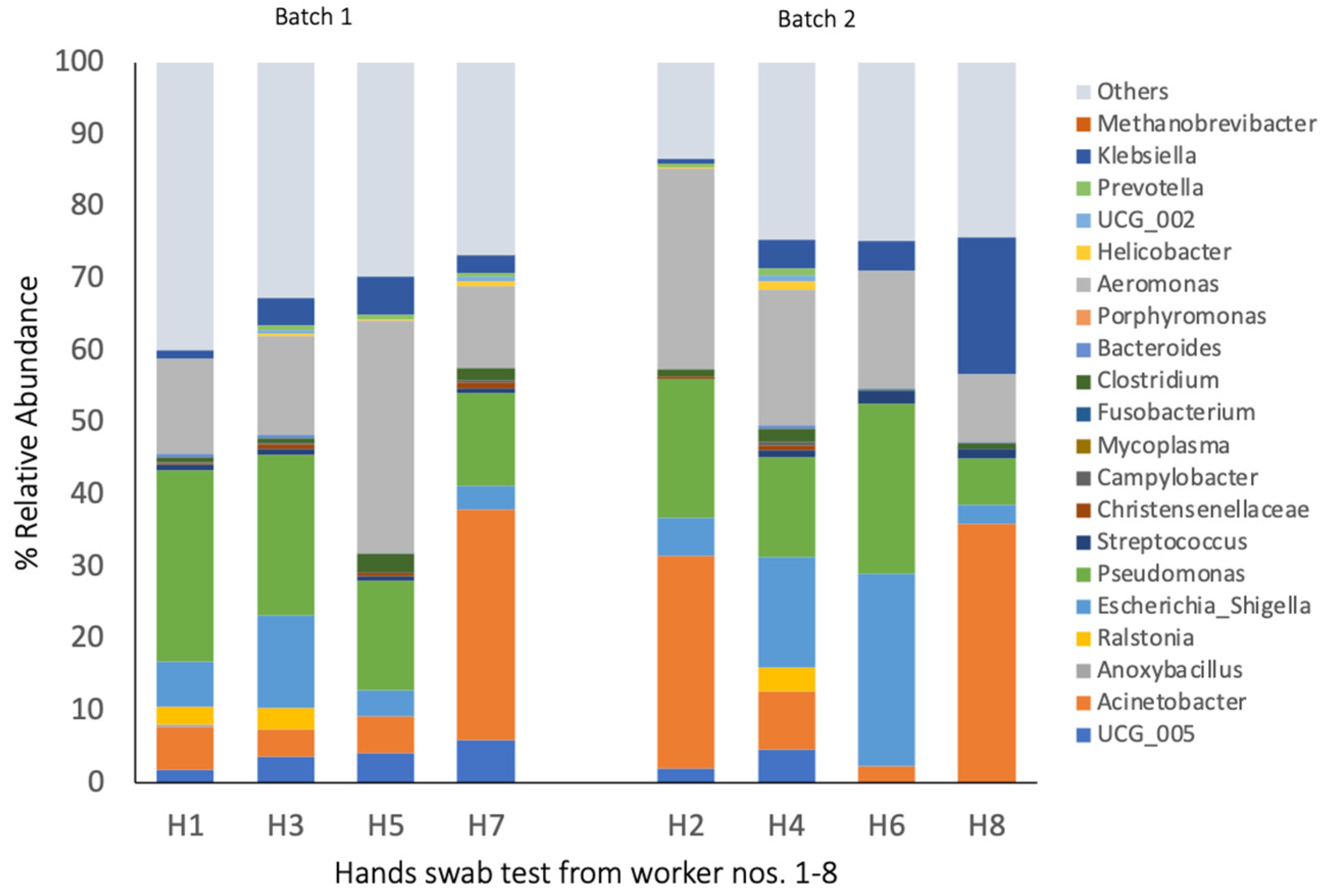
3.4. Bacterial Composition of Hand Swab Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thanapongtharm, W.; Linard, C.; Chinson, P.; Kasemsuwan, S.; Visser, M.; Gaughan, A.E.; Epprech, M.; Robinson, T.P.; Gilbert, M. Spatial analysis and characteristics of pig farming in Thailand. BMC Vet. Res. 2016, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Kang, H.J.; Lee, H.Y.; Jung, H.R.; Moon, J.S.; Yoon, S.S.; Kim, H.Y.; Lee, Y.J. Prevalence and characteristics of foodborne pathogens from slaughtered pig carcasses in Korea. Front. Vet. Sci. 2023, 10, 115819. [Google Scholar] [CrossRef]
- Kittiwan, N.; Tadee, P.; Tadee, P.; Buawiratlert, T.; Eiamsam-Ang, T.; Boonma, O.; Rojanasthien, S.; Pascoe, B.; Patchanee, P.P. Identification of Streptococcus suis carriage in healthy pigs in Chiang Mai, Thailand. Vet. Integr. Sci. 2022, 20, 363–376. [Google Scholar] [CrossRef]
- Asmus, A.E.; Gaire, T.N.; Schweisthal, K.J.; Staben, S.M.; Noyes, N.R. Microbiome characterization of two fresh pork cuts during production in a pork fabrication facility. Microbiol. Spectr. 2025, 30, e0220924. [Google Scholar] [CrossRef]
- Shedleur-Bourguignon, F.; Duchemin, T.; Thériault, W.P.; Longpré, J.; Thibodeau, A.M.; Hocine, N.; Fravalo, P. Distinct microbiotas are associated with different production lines in the cutting room of a swine slaughterhouse. Microorganisms 2023, 11, 133. [Google Scholar] [CrossRef]
- Mann, E.; Dzieciol, M.; Pinior, B.; Neubauer, V.; Metzler-Zebeli, B.U.; Wagner, M.; Schmitz-Esser, S. High Diversity of Viable Bacteria Isolated from Lymph Nodes of Slaughter Pigs and Its Possible Impacts for Food Safety. J. Appl. Microbiol. 2015, 119, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Boonyong, N.; Kaewmongkol, S.; Khunbutsri, D.; Satchasataporn, K.; Meekhanon, N. Contamination of Streptococcus suis in pork and edible pig organs in central Thailand. Vet. World 2019, 12, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Samphao, R.; Chuknam, T.; Rattanathamsakul, T. Epidemiological characteristics of fatal Streptococcus suis infection cases in Thailand from 2016 to 2020. WESR 2022, 53, 141–150. Available online: https://he05.tci-thaijo.org/index.php/WESR/article/view/1021 (accessed on 18 April 2025).
- Gram, L.; Ravn, L.; Rasch, M.; Bruhn, J.B.; Christensen, A.B.; Givskov, M. Food spoilage—Interactions between food spoilage bacteria. Int. J. Food Microbiol. 2002, 78, 79–97. [Google Scholar] [CrossRef]
- Li, J.; Huang, F.; Zhou, Y.; Huang, T.; Tong, X.; Zhang, M.; Chen, J.; Zhang, Z.; Du, H.; Liu, Z.; et al. Comprehensive catalogs for microbial genes and metagenome-assembled genomes of the swine lower respiratory tract microbiome identify the relationship of microbial species with lung lesions. BioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Niazy, M.; Hill, S.; Nadeem, K.; Huber, L.; Harding, J.C.; Slifierz, M.J.; Detmer, S.E.; Weese, J.S. Compositional analysis of the tonsil microbiota in relationship to Streptococcus suis disease in nursery pigs in Ontario. Anim. Microbiome 2022, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Zwirzitz, B.; Wetzels, S.U.; Dixon, E.D.; Stessl, B.; Zaiser, A.; Rabanser, I.; Thalguter, S.; Pinior, B.; Roch, F.F.; Strachan, C.; et al. The sources and transmission routes of microbial populations throughout a meat processing facility. npj Biofilms Microbiomes 2020, 6, 26. [Google Scholar] [CrossRef]
- Ngan, W.Y.; Rao, S.; Chan, L.C.; Sekoai, P.T.; Pu, Y.; Yao, Y.; Fung, A.H.Y.; Habimana, O. Impacts of Wet Market Modernization Levels and Hygiene Practices on the Microbiome and Microbial Safety of Wooden Cutting Boards in Hong Kong. Microorganisms 2020, 8, 1941. [Google Scholar] [CrossRef] [PubMed]
- Asmus, A.E.; Gaire, T.N.; Heimer, K.M.; Belk, K.E.; Singer, R.S.; Johnson, T.J.; Noyes, N.R. Fresh pork microbiota is temporally dynamic and compositionally diverse across meat, contact surfaces, and processing lines in a pork processing facility. Appl. Environ. Microbiol. 2025, 91, e0004425. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Lerch, F.; Yosi, F.; Vötterl, J.; Ehmig, J.; Koger, S.; Verhovsek, D. Temporal Microbial Dynamics in Feces Discriminate by Nutrition, Fecal Color, Consistency and Sample Type in Suckling and Newly Weaned Piglets. Animals 2023, 13, 2251. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package R Package Version 2.5-2. 2018. Available online: https://CRAN.R-project.org/package=vegan (accessed on 15 August 2020).
- Chen, S.H.; Fegan, N.; Kocharunchitt, C.; Bowman, J.P.; Duffy, L.L. Impact of Poultry Processing Operating Parameters on Bacterial Transmission and Persistence on Chicken Carcasses and Their Shelf Life. Appl. Environ. Microbiol. 2020, 86, e00594-20. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Lee, G.Y.; Ali, M.S.; Park, S.C. Effects of dietary intake of heat-inactivated Limosilactobacillus reuteri PSC102 on the growth performance, immune response, and gut microbiota in weaned piglets. Pak. Vet. J. 2024, 44, 819–825. [Google Scholar] [CrossRef]
- Wessels, A.G. Influence of the Gut Microbiome on Feed Intake of Farm Animals. Microorganisms 2022, 10, 1305. [Google Scholar] [CrossRef]
- Viltrop, A.; Niine, T.; Tobias, T.; Sassu, E.L.; Bartolo, I.D.; Pavoni, E.; Smith, R.P. A review of slaughter practices and their effectiveness to control microbial—Esp. Salmonella spp.—Contamination of pig carcasses. J. Food Prot. 2023, 86, 100171. [Google Scholar] [CrossRef]
- Luo, Y.; Ren, W.; Smidt, H.; Wright, A.G.; Yu, B.; Schyns, G.; McCormack, U.M.; Cowieson, A.J.; Yu, J.; He, J.; et al. Dynamic Distribution of Gut Microbiota in Pigs at Different Growth Stages: Composition and Contribution. Microbiol. Spectr. 2022, 10, e00688-21. [Google Scholar] [CrossRef]
- Fredriksen, S.; Guan, X.; Boekhorst, J.; Molist, F.; van Baarlen, P.; Wells, J.M. Environmental and Maternal Factors Shaping Tonsillar Microbiota Development in Piglets. BMC Microbiol. 2022, 22, 224. [Google Scholar] [CrossRef]
- Lyu, C.; Li, Y.; Dong, Y.; Xu, X.; Wang, H. Comprehensive Evaluation of the Bacterial Adhesion and Spoilage Capacity of Meat-Borne Acinetobacter spp. Food Res. Int. 2025, 203, 115831. [Google Scholar] [CrossRef] [PubMed]
- Braley, C.; Fravalo, P.; Gaucher, M.-L.; Larivière-Gauthier, G.; Shedleur-Bourguignon, F.; Longpré, J.; Thibodeau, A. Similar Carcass Surface Microbiota Observed Following Primary Processing of Different Pig Batches. Front. Microbiol. 2022, 13, 849883. [Google Scholar] [CrossRef] [PubMed]
- Lv, R.; Zhang, W.; Sun, Z.; Si, X.; Dong, H.; Liu, X. Current Prevalence and Therapeutic Strategies for Porcine Streptococcus suis in China. Appl. Environ. Microbiol. 2025, 91, e02160-24. [Google Scholar] [CrossRef] [PubMed]
- Holtz, L.R.; Neill, M.A.; Tarr, P.I. Acute Bloody Diarrhea: A Medical Emergency for Patients of All Ages. Gastroenterology 2009, 136, 1887–1898. [Google Scholar] [CrossRef]
- Pena Cortes, L.C.; LeVeque, R.M.; Funk, J.A.; Marsh, T.L.; Mulks, M.H. Development of the Tonsil Microbiome in Pigs and Effects of Stress on the Microbiome. Front. Vet. Sci. 2018, 5, 220. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, H.; Bian, C.; Chen, H.; Shen, Y.; Gao, X.; Ma, J.; Yao, H.; Wang, L.; Wu, Z. The antimicrobial systems of Streptococcus suis promote niche competition in pig tonsils. Virulence 2022, 13, 781–793. [Google Scholar] [CrossRef]
- Wongnak, P.; Wiratsudakul, A.; Nuanualsuwan, S. A risk assessment of pathogenic Streptococcus suis in pork supply chains and markets in Thailand. Food Control 2020, 118, 107432. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Sun, Q.; Wei, J.; Li, B.; Qiu, Y.; Liu, K.; Shao, D.; Ma, Z. Targeting the Pulmonary Microbiota to Fight against Respiratory Diseases. Cells 2022, 11, 916. [Google Scholar] [CrossRef]
- Pirolo, M.; Espinosa-Gongora, C.; Bogaert, D.; Guardabassi, L. The porcine respiratory microbiome: Respiratory Microbiome: Recent Insights and Future Challenges. Anim. Microbiome 2021, 3, 9. [Google Scholar] [CrossRef]
- De Lucia, A.; Ostanello, F. On-farm risk factors associated with Salmonella in pig herds. Large Anim. Rev. 2020, 26, 133–140. Available online: https://vetjournal.it/images/archive/LAR%202020/LAR%203/05%20Ostanello%20imp_ok.pdf (accessed on 30 October 2022).
- André, S.; Vallaeys, T.; Planchon, S. Spore-Forming Bacteria Responsible for Food Spoilage. Res. Microbiol. 2017, 168, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.O.; Lechner, S.L.; Ross, H.C.; Joris, B.R.; Glick, B.R.; Stegelmeier, A.A. Friends and Foes: Bacteria of the Hydroponic Plant Microbiome. Plants 2024, 13, 3069. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, F.M.; Pérez-Wohlfeil, E.; Carvalho, F.M.; Trelles, O.; Schrank, I.S.; Vasconcelos, A.T.R.; Zaha, A. Microbiome overview in swine lungs. PLoS ONE 2017, 12, e0181503. [Google Scholar] [CrossRef]
- Weinroth, M.D.; Belk, A.D.; Dean, C.; Noyes, N.; Dittoe, D.K.; Rothrock, M.J.; Ricke, S.C.; Myer, P.R.; Henniger, M.T.; Ramírez, G.A.; et al. Considerations and Best Practices in Animal Science 16S Ribosomal RNA Gene Sequencing Microbiome Studies. J. Anim. Sci. 2022, 100, skab346. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Index | SEM |
|---|---|---|
| Observed features | ||
| Feces | 551 ab | 45.2 |
| Cecal digesta | 393 bc | 43.0 |
| Cecal mucosa | 635 a | 43.0 |
| Blood | 227 c | 47.6 |
| Tonsil | 370 bc | 43.0 |
| Lung | 284 bc | 50.5 |
| Spleen | 493 ab | 45.2 |
| Meat cut | 381 bc | 53.9 |
| Hand (swab sample) | 249 bc | 62.9 |
| p-value | <0.001 | |
| Shannon | ||
| Feces | 4.67 a | 0.237 |
| Cecal digesta | 5.08 a | 0.225 |
| Cecal mucosa | 5.48 a | 0.225 |
| Blood | 3.08 b | 0.249 |
| Tonsil | 3.93 ab | 0.225 |
| Lung | 2.80 bc | 0.264 |
| Spleen | 2.72 c | 0.237 |
| Meat cut | 4.07 ab | 0.282 |
| Hand (swab sample) | 3.58 bc | 0.330 |
| p-value | <0.001 | |
| Simpson | ||
| Feces | 0.949 a | 0.032 |
| Cecal digesta | 0.980 a | 0.030 |
| Cecal mucosa | 0.986 a | 0.030 |
| Blood | 0.758 b | 0.033 |
| Tonsil | 0.927 a | 0.030 |
| Lung | 0.745 b | 0.035 |
| Spleen | 0.695 b | 0.032 |
| Meat cut | 0.922 ab | 0.038 |
| Hand (Swab sample) | 0.912 ab | 0.044 |
| p-value | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klinsoda, J.; Boonsoongnern, A.; Thanantong, N.; Kaminsonsakul, T.; Treesuwan, K.; Trevanich, S.; Metzler-Zebeli, B.U. The Microbiome Characterization of Edible Visceral Organs and Fresh Meat During Production in a Pig Processing Facility in Thailand. Pathogens 2025, 14, 475. https://doi.org/10.3390/pathogens14050475
Klinsoda J, Boonsoongnern A, Thanantong N, Kaminsonsakul T, Treesuwan K, Trevanich S, Metzler-Zebeli BU. The Microbiome Characterization of Edible Visceral Organs and Fresh Meat During Production in a Pig Processing Facility in Thailand. Pathogens. 2025; 14(5):475. https://doi.org/10.3390/pathogens14050475
Chicago/Turabian StyleKlinsoda, Jutamat, Alongkot Boonsoongnern, Narut Thanantong, Tanyanant Kaminsonsakul, Khemmapas Treesuwan, Sudsai Trevanich, and Barbara U. Metzler-Zebeli. 2025. "The Microbiome Characterization of Edible Visceral Organs and Fresh Meat During Production in a Pig Processing Facility in Thailand" Pathogens 14, no. 5: 475. https://doi.org/10.3390/pathogens14050475
APA StyleKlinsoda, J., Boonsoongnern, A., Thanantong, N., Kaminsonsakul, T., Treesuwan, K., Trevanich, S., & Metzler-Zebeli, B. U. (2025). The Microbiome Characterization of Edible Visceral Organs and Fresh Meat During Production in a Pig Processing Facility in Thailand. Pathogens, 14(5), 475. https://doi.org/10.3390/pathogens14050475