
From Alpha to Delta—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland

 , ,
, ,  , , , , , , and
, , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
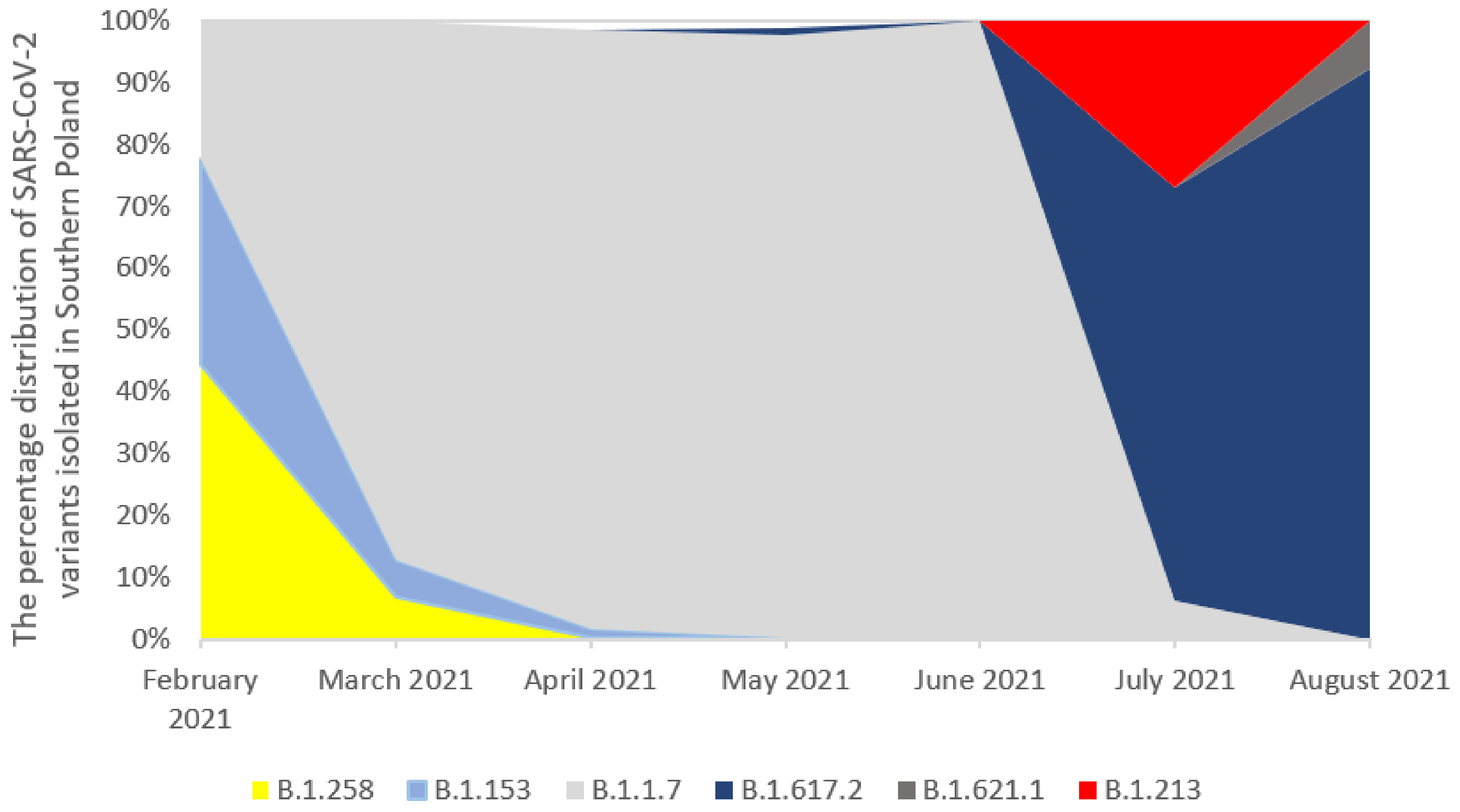
2.1. The Prevalence of SARS-CoV-2 Variants (hCoV-19) in Southern Poland
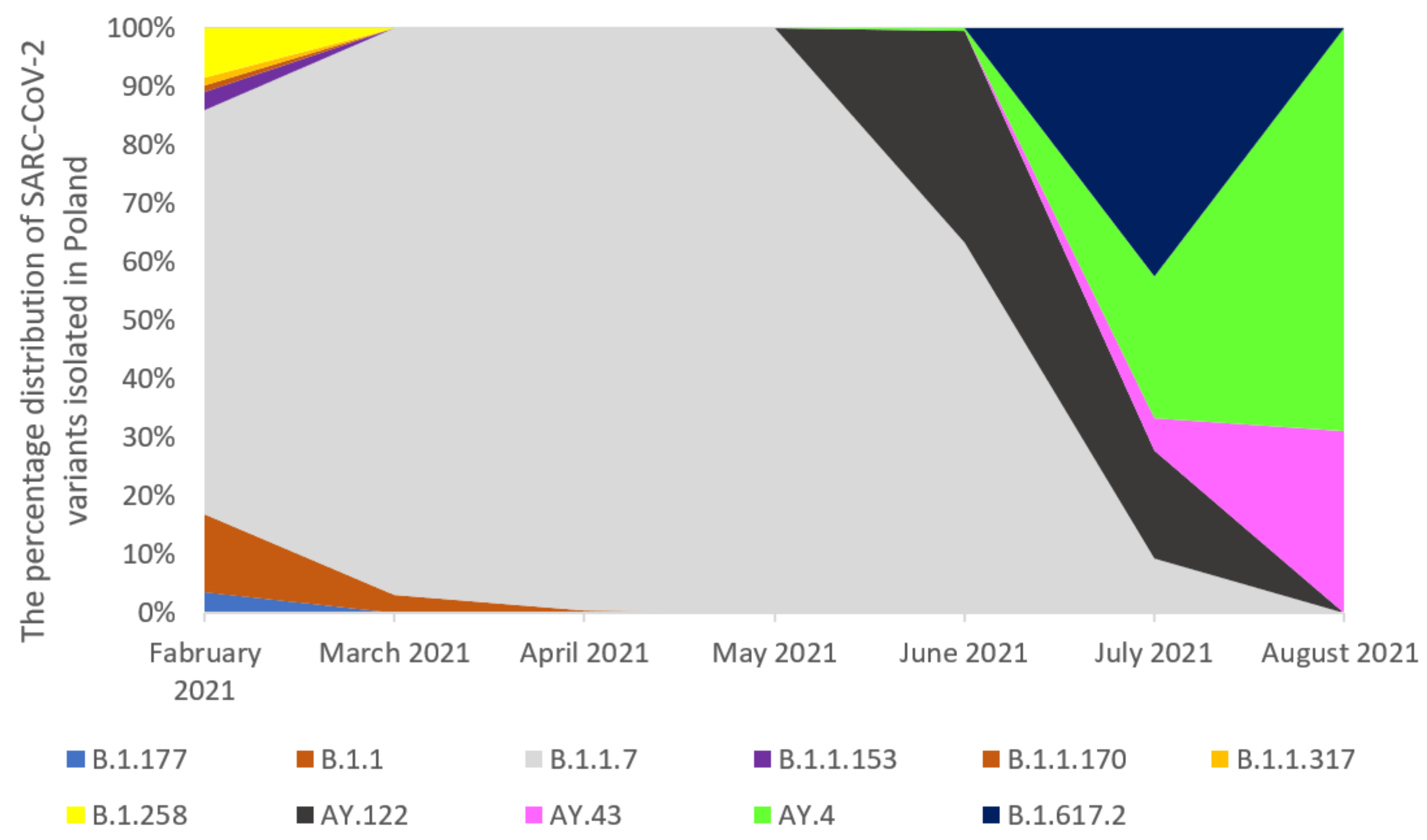
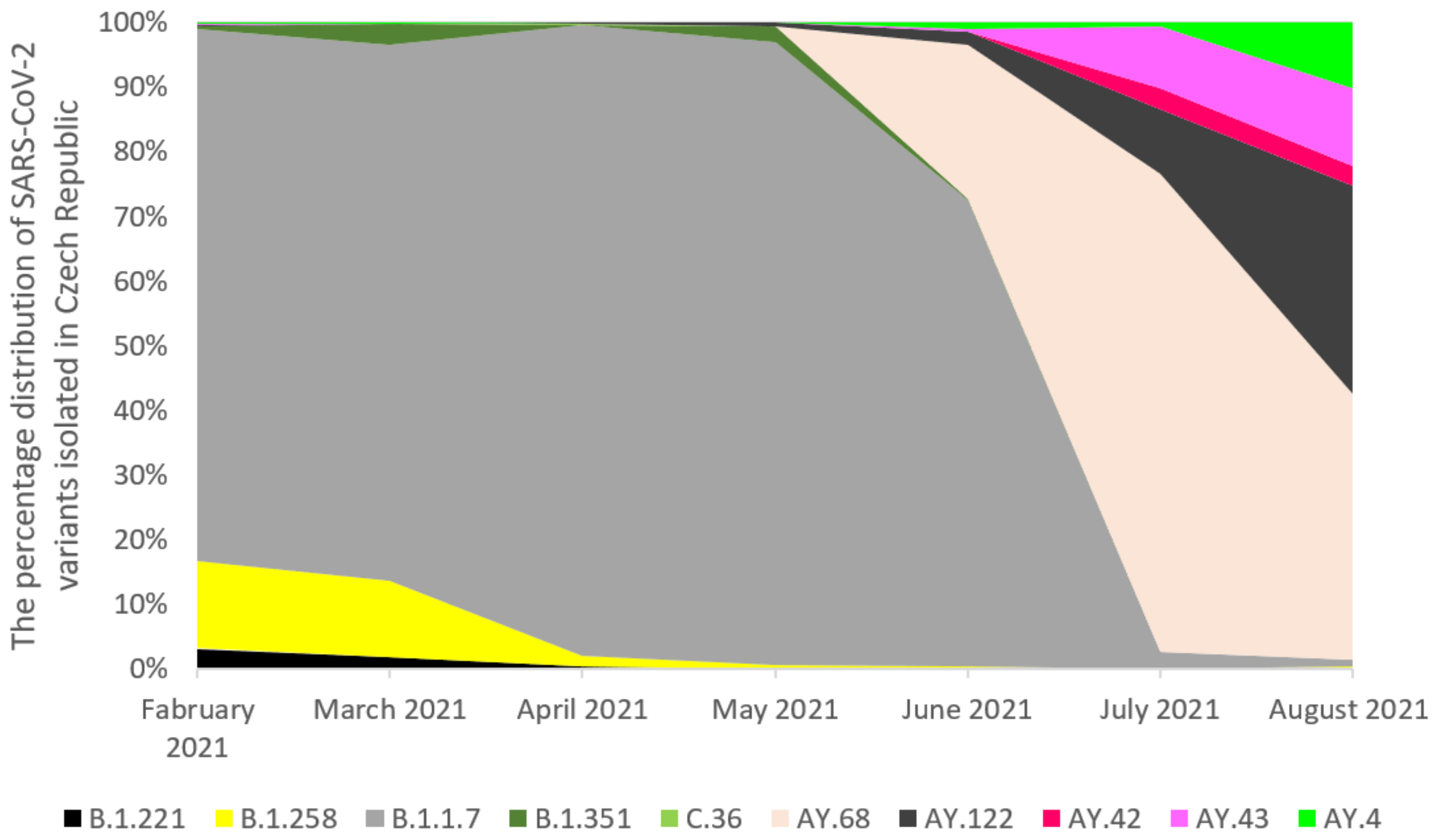
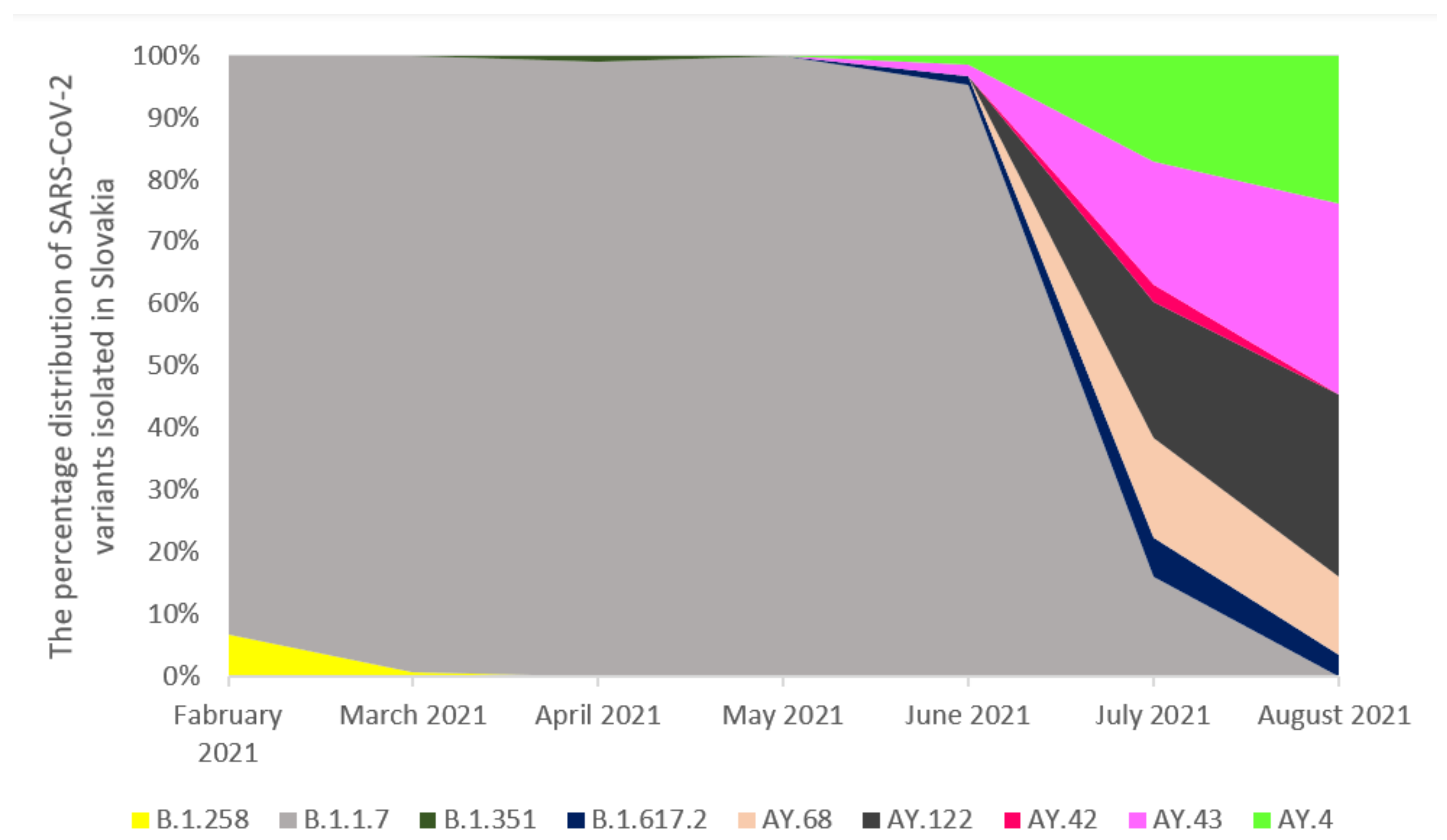
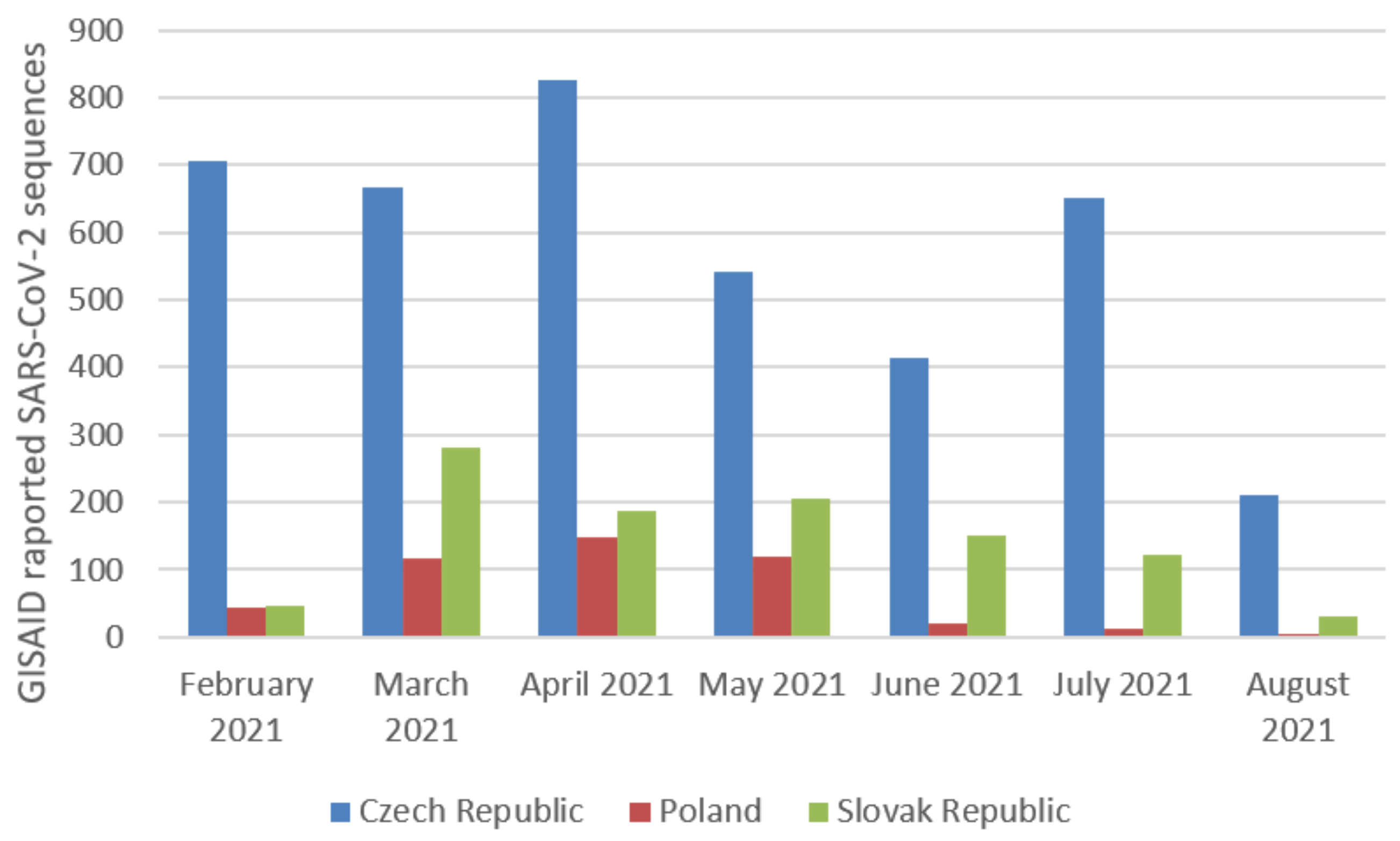
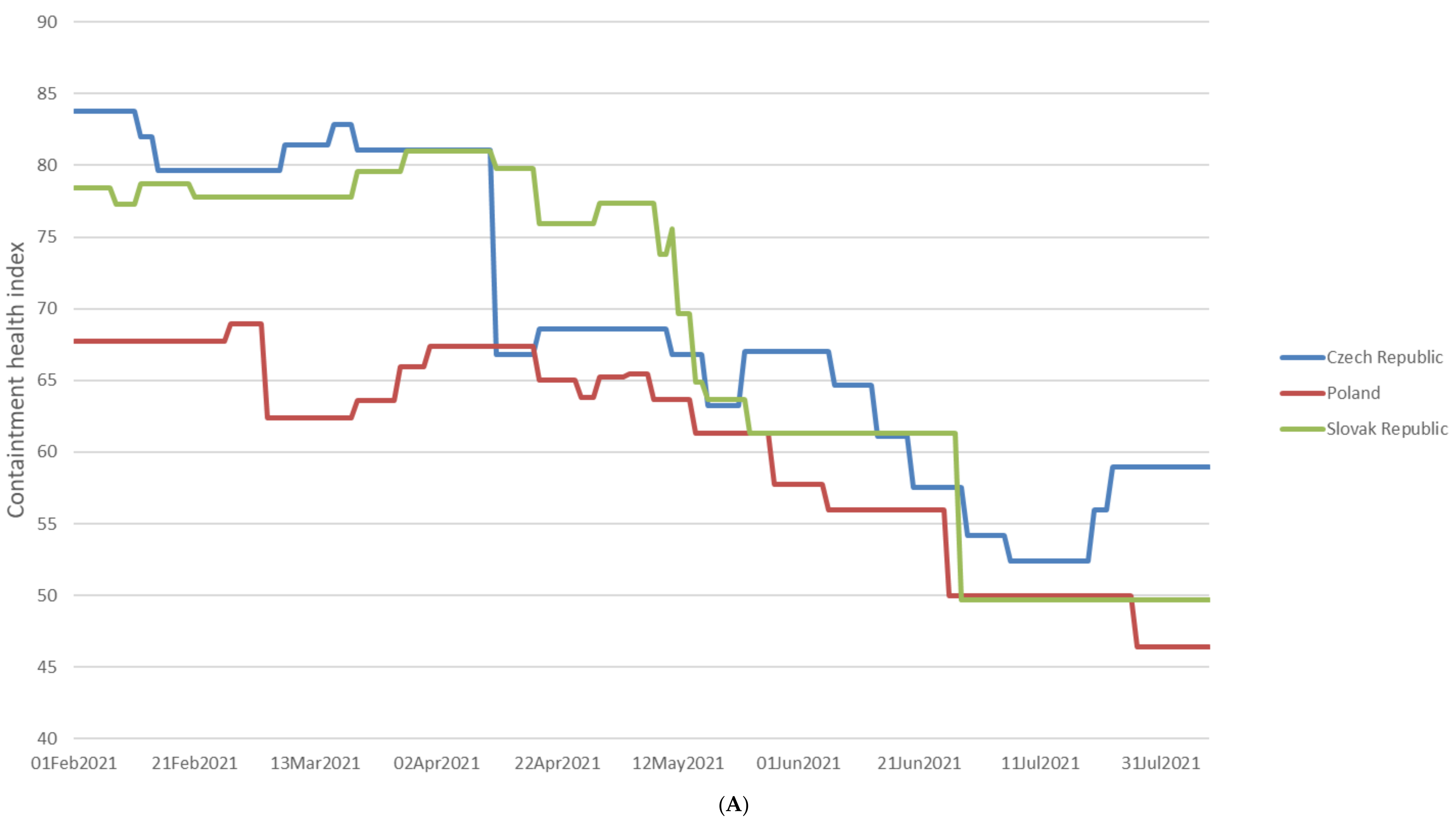
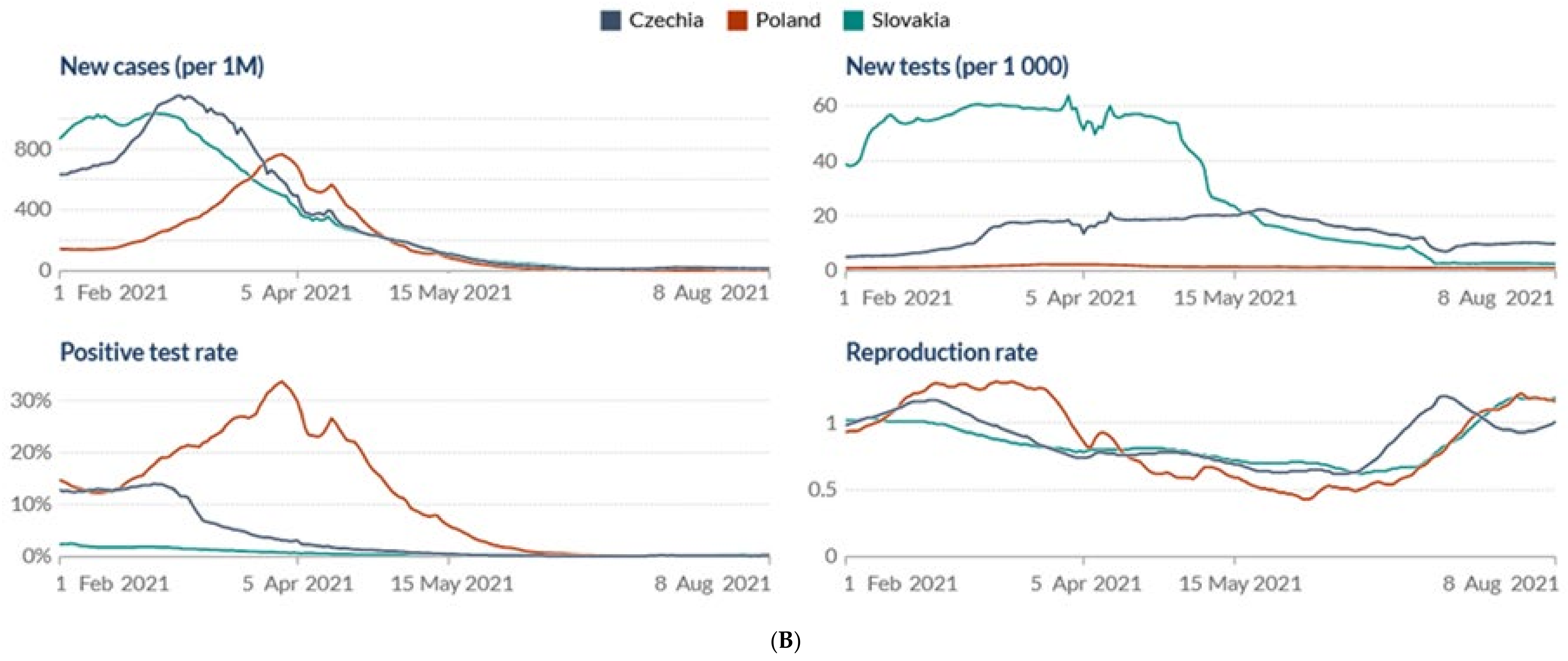
2.2. The Prevalence of SARS-CoV-2 Main Variants (hCoV-19) in Poland, the Czech Republic, and Slovakia
2.3. The Phylogenetic Relationships of SARS-CoV-2 Clades
2.4. Spike Protein Mutations Analysis
3. Discussion
4. Materials and Methods
4.1. Study Group
4.2. SARS-CoV-2 Whole-Genome Sequencing (WGS)
4.3. Analysis of Sequencing Results
4.4. Subsampling of the Data
4.5. Phylogenetic Tree Construction
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Maio, N.; Walker, C.R.; Turakhia, Y.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Mutation Rates and Selection on Synonymous Mutations in SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab087. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Genger, J.W.; Nicholson, M.D.; Penz, T.; Schmid, D.; Aberle, S.W.; Agerer, B.; Lercher, A.; Endler, L.; Colaço, H.; et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 2020, 12, eabe2555. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Nieuwenhuijse, D.F.; Stein, M.; O’Toole, Á.; Haverkate, M.; Mollers, M.; Kamga, S.K.; Schapendonk, C.; Pronk, M.; Lexmond, P.; et al. Rapid SARS-CoV-2 Whole-Genome Sequencing and Analysis for Informed Public Health Decision-Making in the Netherlands. Nat. Med. 2020, 26, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Eurosurveillance 2020, 25, 2001410. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; COVID-19 Genomics UK (COG-UK) Consortium; Network for Genomic Surveillance in South Africa (NGS-SA); Brazil-UK CADDE Genomic Network; et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2 with grinch. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Oxford COVID-19 Government Response Tracker, Blavatnik School of Government, University of Oxford—Last updated 12 June 2022. Available online: OurWorldInData.org/coronavirus (accessed on 12 June 2022).
- Our World in Data. Available online: https://ourworldindata.org/explorers/coronavirus-data-explorer?zoomToSelection=true&time=2021-02-01..2021-0808&uniformYAxis=0&pickerSort=desc&pickerMetric=new_cases_per_million&Metric=Cases%2C+tests%2C+positive+and+reproduction+rate&Interval=7-day+rolling+average&Relative+to+Population=true&Color+by+test+positivity=false&country=POL~CZE~SVK (accessed on 12 June 2022).
- CoVariants. Available online: https://covariants.org/variants/21I.Delta (accessed on 27 January 2022).
- Tasakis, R.N.; Samaras, G.; Jamison, A.; Lee, M.; Paulus, A.; Whitehouse, G.; Verkoczy, L.; Papavasiliou, F.N.; Diaz, M. SARS-CoV-2 Variant Evolution in the United States: High Accumulation of Viral Mutations over Time Likely through Serial Founder Events and Mutational Bursts. PLoS ONE 2021, 16, e0255169. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Serwin, K.; Ossowski, A.; Szargut, M.; Cytacka, S.; Urbańska, A.; Majchrzak, A.; Niedźwiedź, A.; Czerska, E.; Pawińska-Matecka, A.; Gołąb, J.; et al. Molecular Evolution and Epidemiological Characteristics of SARS COV-2 in (Northwestern) Poland. Viruses 2021, 13, 1295. [Google Scholar] [CrossRef] [PubMed]
- Charkiewicz, R.; Nikliński, J.; Biecek, P.; Kiśluk, J.; Pancewicz, S.; Moniuszko-Malinowska, A.M.; Flisiak, R.; Krętowski, A.J.; Dzięcioł, J.; Moniuszko, M.; et al. The First SARS-CoV-2 Genetic Variants of Concern (VOC) in Poland: The Concept of a Comprehensive Approach to Monitoring and Surveillance of Emerging Variants. Adv. Med. Sci. 2021, 66, 237–245. [Google Scholar] [CrossRef]
- Glaß, M.; Misiak, D.; Misiak, C.; Müller, S.; Rausch, A.; Angermann, K.; Hoyer, M.; Zabel, R.; Kehlen, A.; Möbius, B.; et al. Fast forward evolution in real time: The rapid spread of SARS-CoV-2 variant of concern lineage B.1.1.7 in Saxony-Anhalt over a period of 5 months. LaboratoriumsMedizin 2022, 46, 71–75. [Google Scholar] [CrossRef]
- Boršová, K.; Paul, E.D.; Kováčová, V.; Radvánszka, M.; Hajdu, R.; Čabanová, V.; Sláviková, M.; Ličková, M.; Lukáčiková, Ľ.; Belák, A.; et al. Surveillance of SARS-CoV-2 Lineage, B.1.1.7 in Slovakia Using a Novel, Multiplexed RT-QPCR Assay. Sci. Rep. 2021, 11, 20494. [Google Scholar] [CrossRef]
- Klempt, P.; Brzoň, O.; Kašný, M.; Kvapilová, K.; Hubáček, P.; Briksi, A.; Bezdíček, M.; Koudeláková, V.; Lengerová, M.; Hajdúch, M.; et al. Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic. Microorganisms 2021, 9, 1671. [Google Scholar] [CrossRef]
- Goncalves Cabecinhas, A.R.; Roloff, T.; Stange, M.; Bertelli, C.; Huber, M.; Ramette, A.; Chen, C.; Nadeau, S.; Gerth, Y.; Yerly, S.; et al. SARS-CoV-2 N501Y Introductions and Transmissions in Switzerland from Beginning of October 2020 to February 2021-Implementation of Swiss-Wide Diagnostic Screening and Whole Genome Sequencing. Microorganisms 2021, 9, 677. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Klaser, K.; Molteni, E.; Graham, M.; Canas, L.; Osterdahl, M.; Antonelli, M.; Chen, L.; Deng, J.; Murray, B.; Kerfoot, E.; et al. COVID-19 due to the B.1.617.2 variant compared to B.1.1.7 (Alpha) variant of SARS-CoV-2: Two prospective observational cohort studies. medRxiv 2021. [Google Scholar] [CrossRef]
- Available online: https://cov-lineages.org/global_report_B.1.617.2.html (accessed on 27 January 2022).
- Klimczak, L.J.; Randall, T.A.; Saini, N.; Li, J.L.; Gordenin, D.A. Similarity between Mutation Spectra in Hypermutated Genomes of Rubella Virus and in SARS-CoV-2 Genomes Accumulated during the COVID-19 Pandemic. PLoS ONE 2020, 15, e0237689. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Nandi, S.; Saha, I. A Review on Evolution of Emerging SARS-CoV-2 Variants Based on Spike Glycoprotein. Int. Immunopharmacol. 2022, 105, 108565. [Google Scholar] [CrossRef]
- Jia, Z.; Gong, W. Will Mutations in the Spike Protein of SARS-CoV-2 Lead to the Failure of COVID-19 Vaccines? J. Korean Med. Sci. 2021, 26, 1–11. [Google Scholar] [CrossRef]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L. Novel SARS-CoV-2 Variants: The Pandemics within the Pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, X.; Chang, C.; Chen, D.; Hao, Y. What is the relationship between government response and COVID-19 pandemics? Global evidence of 118 countries? Struct. Chang. Econ. Dyn. 2021, 59, 98–107. [Google Scholar] [CrossRef]
- Ma, Y.; Mishra, S.R.; Han, X.K.; Zhu, D.S. The Relationship between Time to a High COVID-19 Response Level and Timing of Peak Daily Incidence: An Analysis of Governments’ Stringency Index from 148 Countries. Infect. Dis. Poverty 2021, 10, 96. [Google Scholar] [CrossRef]
- Available online: https://github.com/W-L/ProblematicSites_SARS-CoV2 (accessed on 27 January 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morawiec, E.; Miklasińska-Majdanik, M.; Bratosiewicz-Wąsik, J.; Wojtyczka, R.D.; Swolana, D.; Stolarek, I.; Czerwiński, M.; Skubis-Sikora, A.; Samul, M.; Polak, A.; et al. From Alpha to Delta—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens 2022, 11, 780. https://doi.org/10.3390/pathogens11070780
Morawiec E, Miklasińska-Majdanik M, Bratosiewicz-Wąsik J, Wojtyczka RD, Swolana D, Stolarek I, Czerwiński M, Skubis-Sikora A, Samul M, Polak A, et al. From Alpha to Delta—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens. 2022; 11(7):780. https://doi.org/10.3390/pathogens11070780
Chicago/Turabian StyleMorawiec, Emilia, Maria Miklasińska-Majdanik, Jolanta Bratosiewicz-Wąsik, Robert D. Wojtyczka, Denis Swolana, Ireneusz Stolarek, Michał Czerwiński, Aleksandra Skubis-Sikora, Magdalena Samul, Agnieszka Polak, and et al. 2022. "From Alpha to Delta—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland" Pathogens 11, no. 7: 780. https://doi.org/10.3390/pathogens11070780
APA StyleMorawiec, E., Miklasińska-Majdanik, M., Bratosiewicz-Wąsik, J., Wojtyczka, R. D., Swolana, D., Stolarek, I., Czerwiński, M., Skubis-Sikora, A., Samul, M., Polak, A., Kruszniewska-Rajs, C., Pudełko, A., Figlerowicz, M., Bednarska-Czerwińska, A., & Wąsik, T. J. (2022). From Alpha to Delta—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens, 11(7), 780. https://doi.org/10.3390/pathogens11070780