
Metabolic Control by DNA Tumor Virus-Encoded Proteins



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. MYC Metabolic Targets
1.2. p53 Metabolic Targets
1.3. pRb/E2F Metabolic Targets
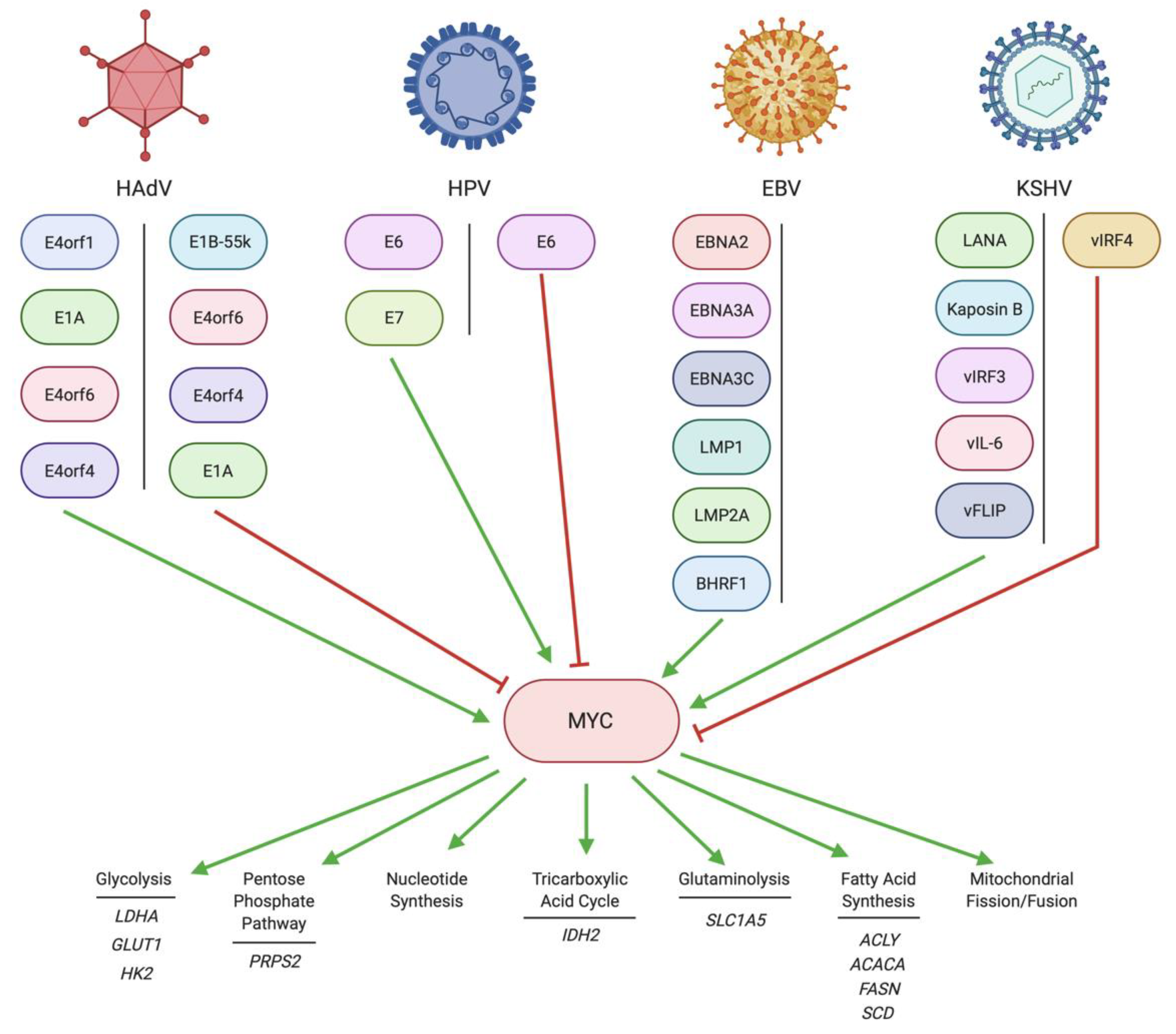
2. Regulation of MYC by DNA Tumor Virus Oncoproteins
2.1. Regulation of MYC by HAdV
2.2. Regulation of MYC by HPV
2.3. Regulation of MYC by EBV
2.4. Regulation of MYC by KSHV
3. Regulation of p53 by DNA Tumor Virus Oncoproteins
3.1. Regulation of p53 by HAdV
3.2. Regulation of p53 by HPV
3.3. Regulation of p53 by EBV
3.4. Regulation of p53 by KSHV
4. Regulation of pRb/E2F by DNA Tumor Virus Oncoproteins
4.1. Regulation of pRb/E2F by HAdV
4.2. Regulation of pRb/E2F by HPV
4.3. Regulation of pRb/E2F by EBV
4.4. Regulation of pRb/E2F by KSHV
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. c-MYC programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-MYC transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor MYC Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.L.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-MYC. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-MYC. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef]
- Zhao, X.; Petrashen, A.P.; Sanders, J.A.; Peterson, A.L.; Sedivy, J.M. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived MYC hypomorphic mice. Aging Cell 2019, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Morrish, F.; Isern, N.; Sadilek, M.; Jeffrey, M.; Hockenbery, D.M. C-MYC activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene 2009, 28, 2485–2491. [Google Scholar] [CrossRef]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef]
- Morrish, F.; Noonan, J.; Perez-Olsen, C.; Gafken, P.R.; Fitzgibbon, M.; Kelleher, J.; VanGilst, M.; Hockenbery, D. MYC-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J. Biol. Chem. 2010, 285, 36267–36274. [Google Scholar] [CrossRef]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; van Houten, B.; Vockley, J.; et al. Mitochondrial structure, function and dynamics are temporally controlled by c-MYC. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC deregulation in primary human cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate MYC protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates MYC-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Furuhashi, M.; Aoki, H.; Goto, D.; Kuwano, H.; Sugamura, K.; Miyazono, K.; Kato, M. c-MYC is a downstream target of the Smad pathway. J. Biol. Chem. 2002, 277, 854–861. [Google Scholar] [CrossRef]
- Lord, J.D.; McIntosh, B.C.; Greenberg, P.D.; Nelson, B.H. The IL-2 Receptor Promotes Lymphocyte Proliferation and Induction of the c- MYC, bcl-2, and bcl-x Genes Through the trans- Activation Domain of Stat5. J. Immunol. 2000, 164, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Dubik, D.; Shiu, R.P. Mechanism of estrogen activation of c-MYC oncogene expression. Oncogene 1992, 7, 1587–1594. [Google Scholar]
- Yochum, G.S. Multiple wnt/ß-catenin responsive enhancers align with the MYC promoter through long-range chromatin loops. PLoS ONE 2011, 6, e18966. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934. [Google Scholar] [CrossRef]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; O’Connor, P.M.; Fornace, A.J. Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene 1995, 10, 1053–1059. [Google Scholar] [PubMed]
- Chandel, N.S.; Vander Heiden, M.G.; Thompson, C.B.; Schumacker, P.T. Redox regulation of p53 during hypoxia. Oncogene 2000, 19, 3840–3848. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Xu, H.; Kufe, D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer Cell 2005, 7, 167–178. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Coates, P.J.; Save, V.; Ansari, B.; Hall, P.A. Demonstration of DNA damage/repair in individual cells using in situ end labelling: Association of p53 with sites of DNA damage. J. Pathol. 1995, 176, 19–26. [Google Scholar] [CrossRef]
- Yonish-Rouach, E.; Resnftzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, L.R.; White, A.; Sprouse, J.; Livanos, E.; Jacks, T.; Tlsty, T.D. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 1992, 70, 923–935. [Google Scholar] [CrossRef]
- Haupt, Y.; Mayat, R.; Kazazt, A.; Orent, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kapoor, M.; Lozano, G. Functional activation of p53 via phosphorylation following DNA damage by UV but not γ radiation. Proc. Natl. Acad. Sci. USA 1998, 95, 2834–2837. [Google Scholar] [CrossRef]
- Tang, Q.; Su, Z.; Gu, W.; Rustgi, A.K. Mutant p53 on the Path to Metastasis. Trends Cancer 2020, 6, 62–73. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Bellacchio, E.; Paggi, M.G. Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J. Cell. Physiol. 2013, 228, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Sdek, P.; Ying, H.; Chang, D.L.F.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Jim Xiao, Z.X. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 2005, 20, 699–708. [Google Scholar] [CrossRef]
- Medema, R.H.; Herrera, R.E.; Lam, F.; Weinberg, R.A. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc. Natl. Acad. Sci. USA 1995, 92, 6289–6293. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Ludlow, J.W.; Lynch, D.; Furukawa, Y.; Griffin, J.; Piwnica-Worms, H.; Huang, C.M.; Livingston, D.M. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 1989, 58, 1085–1095. [Google Scholar] [CrossRef]
- Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Demonacos, C.; La Thangue, N.B. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 2001, 3, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Santockyte, R.; Shen, R.F.; Tekle, E.; Wang, G.; Yang, D.C.H.; Chock, P.B. Expression of SUMO-2/3 induced senescence through p53- and pRB-mediated pathways. J. Biol. Chem. 2006, 281, 36221–36227. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ghosh, M.K. HAUSP, a novel deubiquitinase for Rb - MDM2 the critical regulator. FEBS J. 2014, 281, 3061–3078. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 2013, 25, 735–740. [Google Scholar] [CrossRef]
- Darville, M.I.; Antoine, I.V.; Mertens-Strijthagen, J.R.; Dupriez, V.J.; Rousseau, G.G. An E2F-dependent late-serum-response promoter in a gene that controls glycolysis. Oncogene 1995, 11, 1509–1517. [Google Scholar]
- De Mattos, S.F.; Lam, E.W.F.; Tauler, A. An E2F-binding site mediates the activation of the proliferative isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase by phosphatidylinositol 3-kinase. Biochem. J. 2002, 368, 283–291. [Google Scholar] [CrossRef][Green Version]
- Hsieh, M.C.F.; Das, D.; Sambandam, N.; Zhang, M.Q.; Nahlé, Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J. Biol. Chem. 2008, 283, 27410–27417. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Gameiro, P.A.; Tschöp, K.; Korenjak, M.; Heilmann, A.M.; Asara, J.M.; Stephanopoulos, G.; Iliopoulos, O.; Dyson, N.J. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013, 27, 182–196. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Functions of adenovirus E1A. Cancer Surv. 1986, 5, 367–387. [Google Scholar]
- Barker, D.D.; Berk, A.J. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 1987, 156, 107–121. [Google Scholar] [CrossRef]
- Javier, R.T. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J. Virol. 1994, 68, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Nevels, M.; Rubenwolf, S.; Spruss, T.; Wolf, H.; Dobner, T. Two Distinct Activities Contribute to the Oncogenic Potential of the Adenovirus Type 5 E4orf6 Protein. J. Virol. 2000, 74, 5168–5181. [Google Scholar] [CrossRef]
- Valdés, A.; Zhao, H.; Pettersson, U.; Lind, S.B. Time-resolved proteomics of adenovirus infected cells. PLoS ONE 2018, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.; Kumar, M.; Taruishi, M.; Javier, R.T. Adenovirus E4-ORF1 Dysregulates Epidermal Growth Factor and Insulin/Insulin-Like Growth Factor Receptors To Mediate Constitutive MYC Expression. J. Virol. 2015, 89, 10774–10785. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Loewenstein, P.M.; Green, M. Enhanced MYC association with the NuA4 histone acetyltransferase complex mediated by the adenovirus E1A N-terminal domain activates a subset of MYC target genes highly expressed in cancer cells. Genes Cancer 2017, 8, 752. [Google Scholar] [CrossRef]
- Löhr, K.; Hartmann, O.; Schäfer, H.; Dobbelstein, M. Mutual Interference of Adenovirus Infection and MYC Expression. J. Virol. 2003, 77, 7936–7944. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tworkowski, K.A.; Chakraborty, A.A.; Samuelson, A.V.; Seger, Y.R.; Narita, M.; Hannon, G.J.; Lowe, S.W.; Tansey, W.P. Adenovirus E1A targets p400 to induce the cellular oncoprotein MYC. Proc. Natl. Acad. Sci. USA 2008, 105, 6103–6108. [Google Scholar] [CrossRef]
- Kadeppagari, R.-K.; Sankar, N.; Thimmapaya, B. Adenovirus Transforming Protein E1A Induces c-MYC in Quiescent Cells by a Novel Mechanism. J. Virol. 2009, 83, 4810–4822. [Google Scholar] [CrossRef] [PubMed]
- Prusinkiewicz, M.A.; Tu, J.; Dodge, M.J.; MacNeil, K.M.; Radko-Juettner, S.; Fonseca, G.J.; Pelka, P.; Mymryk, J.S. Differential effects of human adenovirus E1A protein isoforms on aerobic glycolysis in A549 human lung epithelial cells. Viruses 2020, 12, 610. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-J.; Loewenstein, P.M.; Green, M. The adenoviral E1A N-terminal domain represses MYC transcription in human cancer cells by targeting both p300 and TRRAP and inhibiting MYC promoter acetylation of H3K18 and H4K16. Genes Cancer 2016, 7, 98–109. [Google Scholar] [CrossRef]
- Higashino, F.; Aoyagi, M.; Takahashi, A.; Ishino, M.; Taoka, M.; Isobe, T.; Kobayashi, M.; Totsuka, Y.; Kohgo, T.; Shindoh, M. Adenovirus E4orf6 targets pp32/LANP to control the fate of ARE-containing mRNAs by perturbing the CRM1-dependent mechanism. J. Cell Biol. 2005, 170, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ben-Israel, H.; Sharf, R.; Rechavi, G.; Kleinberger, T. Adenovirus E4orf4 Protein Downregulates MYC Expression through Interaction with the PP2A-B55 Subunit. J. Virol. 2008, 82, 9381–9388. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol 1977, 36, 59–66. [Google Scholar] [CrossRef]
- Arnold, H.K.; Sears, R.C. Protein Phosphatase 2A Regulatory Subunit B56α Associates with c-MYC and Negatively Regulates c-MYC Accumulation. Mol. Cell. Biol. 2006, 26, 2832–2844. [Google Scholar] [CrossRef]
- Prusinkiewicz, M.A.; Mymryk, J.S. Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection. Viruses 2019, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Peta, E.; Sinigaglia, A.; Masi, G.; Di Camillo, B.; Grassi, A.; Trevisan, M.; Messa, L.; Loregian, A.; Manfrin, E.; Brunelli, M.; et al. HPV16 E6 and E7 upregulate the histone lysine demethylase KDM2B through the c-MYC/miR-146a-5p axys. Oncogene 2018, 37, 1654–1668. [Google Scholar] [CrossRef]
- Zhang, Y.; Dakic, A.; Chen, R.; Dai, Y.; Schlegel, R.; Liu, X. Direct HPV E6/MYC interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget 2017, 8, 96323–96339. [Google Scholar] [CrossRef]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and MYC proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase Activation by Human Papillomavirus Type 16 E6 Protein: Induction of Human Telomerase Reverse Transcriptase Expression through MYC and GC-Rich Sp1 Binding Sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef]
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and human papillomavirus E6 oncoprotein-induced degradation of MYC proteins by the ubiquitin pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063. [Google Scholar] [CrossRef]
- Liu, X.; Disbrow, G.L.; Yuan, H.; Tomaić, V.; Schlegel, R. MYC and Human Papillomavirus Type 16 E7 Genes Cooperate To Immortalize Human Keratinocytes. J. Virol. 2007, 81, 12689–12695. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Chang, H.S.; Lin, C.H.; Yu, W.C.Y. HPV-18 E7 conjugates to c-MYC and mediates its transcriptional activity. Int. J. Biochem. Cell Biol. 2007, 39, 402–412. [Google Scholar] [CrossRef]
- Strickland, S.W.; Vande Pol, S. The Human Papillomavirus 16 E7 Oncoprotein Attenuates AKT Signaling To Promote Internal Ribosome Entry Site-Dependent Translation and Expression of c-MYC. J. Virol. 2016, 90, 5611–5621. [Google Scholar] [CrossRef]
- Morandell, D.; Kaiser, A.; Herold, S.; Rostek, U.; Lechner, S.; Mitterberger, M.C.; Jansen-Dürr, P.; Eilers, M.; Zwerschke, W. The human papillomavirus type 16 E7 oncoprotein targets MYC-interacting zinc-finger protein-1. Virology 2012, 422, 242–253. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Roy, A.; Roy, B.; Roychoudhury, S.; Panda, C.K. MYC gene amplification reveals clinical association with head and neck squamous cell carcinoma in Indian patients. J. Oral Pathol. Med. 2009, 38, 759–763. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Sabbir, M.G.; Roy, A.; Dam, A.; Roychoudhury, S.; Panda, C.K. Approximately 580 Kb surrounding the MYC gene is amplified in head and neck squamous cell carcinoma of Indian patients. Pathol. Res. Pract. 2005, 201, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Koo, B.S.; Kang, S.; Park, K.; Kim, H.; Lee, K.R.; Lee, M.J.; Kim, J.M.; Choi, E.C.; Cho, N.H. HPV integration begins in the tonsillar crypt and leads to the alteration of p16, EGFR and c-MYC during tumor formation. Int. J. Cancer 2007, 120, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Arizmendi-Izazaga, A.; Navarro-Tito, N.; Jiménez-Wences, H.; Mendoza-Catalán, M.A.; Martínez-Carrillo, D.N.; Zacapala-Gómez, A.E.; Olea-Flores, M.; Dircio-Maldonado, R.; Torres-Rojas, F.I.; Soto-Flores, D.G.; et al. Metabolic Reprogramming in Cancer: Role of HPV 16 Variants. Pathogens 2021, 10, 347. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-MYC onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J. How does Epstein-Barr virus (EBV) complement the activation of MYC in the pathogenesis of Burkitt’s lymphoma? Semin. Cancer Biol. 2009, 19, 366–376. [Google Scholar] [CrossRef]
- Fitzsimmons, L.; Kelly, G.L. EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses 2017, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The Proto-Oncogene c-MYC Is a Direct Target Gene of Epstein-Barr Virus Nuclear Antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.W.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife 2016, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Schmidt, S.C.S.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr Virus Oncoprotein Super-enhancers Control B Cell Growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, H.; Gerdt, C.; Tan, M.; Colson, T.; Kaye, K.M.; Kieff, E.; Zhao, B. Epstein-Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 14121–14126. [Google Scholar] [CrossRef]
- Wang, L.W.; Shen, H.; Nobre, L.; Ersing, I.; Paulo, J.A.; Trudeau, S.; Wang, Z.; Smith, N.A.; Ma, Y.; Reinstadler, B.; et al. Epstein-Barr-Virus-Induced One-Carbon Metabolism Drives B Cell Transformation. Cell Metab. 2019, 30, 539–555.e11. [Google Scholar] [CrossRef]
- Wang, L.W.; Wang, Z.; Ersing, I.; Nobre, L.; Guo, R.; Jiang, S.; Trudeau, S.; Zhao, B.; Weekes, M.P.; Gewurz, B.E. Epstein-barr virus subverts mevalonate and fatty acid pathways to promote infected B-cell proliferation and survival. PLoS Pathog. 2019, 15, e1008030. [Google Scholar] [CrossRef]
- Magon, K.L.; Parish, J.L. From infection to cancer: How DNA tumour viruses alter host cell central carbon and lipid metabolism. Open Biol. 2021, 11, 210004. [Google Scholar] [CrossRef]
- Jiang, S.; Zhou, H.; Liang, J.; Gerdt, C.; Wang, C.; Ke, L.; Schmidt, S.C.S.; Narita, Y.; Ma, Y.; Wang, S.; et al. The Epstein-Barr Virus Regulome in Lymphoblastoid Cells. Cell Host Microbe 2017, 22, 561–573.e4. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, B.G.; Murakami, M.; Cai, Q.; Verma, S.C.; Lan, K.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Interacts with and Enhances the Stability of the c-MYC Oncoprotein. J. Virol. 2008, 82, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Styles, C.T.; Paschos, K.; White, R.E.; Farrell, P.J. The cooperative functions of the EBNA3 proteins are central to EBV persistence and latency. Pathogens 2018, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Dirmeier, U.; Hoffmann, R.; Kilger, E.; Schultheiss, U.; Briseño, C.; Gires, O.; Kieser, A.; Eick, D.; Sugden, B.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 2005, 24, 1711–1717. [Google Scholar] [CrossRef]
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A Positive Autoregulatory Loop of LMP1 Expression and STAT Activation in Epithelial Cells Latently Infected with Epstein-Barr Virus. J. Virol. 2003, 77, 4139–4148. [Google Scholar] [CrossRef]
- Kwok Fung Lo, A.; Wai Lo, K.; Tsao, S.W.; Wong, H.L.; Ying Hui, J.W.; To, K.F.; Hayward, S.D.; Chui, Y.L.; Lau, Y.L.; Takada, K.; et al. Epstein-Barr Virus Infection Alters Cellular Signal Cascades in Human Nasopharyngeal Epithelial Cells. Neoplasia 2006, 8, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; He, Y.; Li, J.; Tang, M.; Li, Y.; Xie, L.; Zhao, L.; Hu, J.; Luo, X.; Zhou, M.; et al. Wild-type IDH2 contributes to Epstein–Barr virus-dependent metabolic alterations and tumorigenesis. Mol. Metab. 2020, 36, 1–11. [Google Scholar] [CrossRef]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X.; et al. Targeting the signaling in Epstein–Barr virus-associated diseases: Mechanism, regulation, and clinical study. Signal Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef]
- Shin, D.Y.; Kim, A.; Kang, H.J.; Park, S.; Kim, D.W.; Lee, S.S. Histone deacetylase inhibitor romidepsin induces efficient tumor cell lysis via selective down-regulation of LMP1 and c-MYC expression in EBV-positive diffuse large B-cell lymphoma. Cancer Lett. 2015, 364, 89–97. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1α by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Fish, K.; Sora, R.P.; Schaller, S.J.; Longnecker, R.; Ikeda, M. EBV latent membrane protein 2A orchestrates p27kip1 degradation via Cks1 to accelerate MYC-driven lymphoma in mice. Blood 2017, 130, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Scott, R.S.; Amirghahari, N.; Nathan, C.-A.; Young, L.S.; Dawson, C.W.; Sixbey, J.W. Modulation of the Cell Growth Regulator mTOR by Epstein-Barr Virus-Encoded LMP2A. J. Virol. 2005, 79, 5499–5506. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, L.; Cartlidge, R.; Chang, C.; Sejic, N.; Galbraith, L.C.A.; Suraweera, C.D.; Grant, D.C.; Tierney, R.J.; Bell, A.I.; Herold, C.S.M.J.; et al. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020, 1554–1568. [Google Scholar] [CrossRef]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 78, 653–669.e8. [Google Scholar] [CrossRef]
- Ishii, H.; Gobé, G.; Kawakubo, Y.; Sato, Y.; Ebihara, Y. Interrelationship between Epstein-Barr virus infection in gastric carcinomas and the expression of apoptosis-associated proteins. Histopathology 2001, 38, 111–119. [Google Scholar] [CrossRef]
- Lima, V.P.; de Lima, M.A.P.; André, A.R.; Ferreira, M.V.P.; Barros, M.A.P.; Rabenhorst, S.H.B. H pylori (CagA) and Epstein-Barr virus infection in gastric carcinomas: Correlation With p53 mutation and c-MYC, Bcl-2 and Bax expression. World J. Gastroenterol. 2008, 14, 884–891. [Google Scholar] [CrossRef]
- Lima, M.A.P.; Ferreira, M.V.P.; Barros, M.A.P.; Pardini, M.I.M.C.; Ferrasi, A.C.; Mota, R.M.S.; Rabenhorst, S.H.B. Relationship Between EBV Infection and Expression of Cellular Proteins c-MYC, Bcl-2, and Bax in Gastric Carcinomas. Diagnostic Mol. Pathol. 2008, 17, 82–89. [Google Scholar] [CrossRef]
- Zhu, S.; Sun, P.; Zhang, Y.; Yan, L.; Luo, B. Expression of c-MYC and PCNA in Epstein-Barr virus-associated gastric carcinoma. Exp. Ther. Med. 2013, 5, 1030–1034. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luo, B.; Wang, Y.; Wang, X.F.; Gao, Y.; Huang, B.H.; Zhao, P. Correlation of Epstein-Barr virus and its encoded proteins with Helicobacter pylori and expression of c-met and c-MYC in gastric carcinoma. World J. Gastroenterol. 2006, 12, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Purdy, J.G.; Luftig, M.A. Reprogramming of cellular metabolic pathways by human oncogenic viruses. Curr. Opin. Virol. 2019, 39, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K. KSHV genome replication and maintenance in latency. In Human Herpesviruses; Kawaguchi, Y., Mori, Y., Kimura, H., Eds.; Springer Singapore Pte. Limited: Singapore, 2018; pp. 299–320. ISBN 9789811072307. [Google Scholar]
- Lo, A.K.-F.; Dawson, C.W.; Young, L.S.; Lo, K.-W. The role of metabolic reprogramming in γ-herpesvirus-associated oncogenesis. Int. J. Cancer 2017, 141, 1512–1521. [Google Scholar] [CrossRef]
- Liu, J.; Martin, H.J.; Liao, G.; Hayward, S.D. The Kaposi’s Sarcoma-Associated Herpesvirus LANA Protein Stabilizes and Activates c-MYC. J. Virol. 2007, 81, 10451–10459. [Google Scholar] [CrossRef]
- Fujimuro, M.; Wu, F.Y.; Aprhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of β-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Bubman, D.; Guasparri, I.; Cesarman, E. Deregulation of c-MYC in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 2007, 26, 4979–4986. [Google Scholar] [CrossRef]
- Chang, H.C.; Hsieh, T.H.; Lee, Y.W.; Tsai, C.F.; Tsai, Y.N.; Cheng, C.C.; Wang, H.W. c-MYC and viral cofactor Kaposin B co-operate to elicit angiogenesis through modulating miRNome traits of endothelial cells. BMC Syst. Biol. 2016, 10, 1–16. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Mele, J.; Guo, C.; Myatt, L.G.; Myatt, L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 2012, 33, 816–823. [Google Scholar] [CrossRef]
- Ma, M.; Ma, C.; Li, P.; Ma, C.; Ping, F.; Li, W.; Xu, L.; Zhang, H.; Sun, Q.; Li, Y. Low glucose enhanced metformin’s inhibitory effect on pancreatic cancer cells by suppressing glycolysis and inducing energy stress via up-regulation of miR-210-5p. Cell Cycle 2020, 19, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Pranzini, E.; Leo, A.; Rapizzi, E.; Ramazzotti, M.; Magherini, F.; Giovannelli, L.; Caselli, A.; Cirri, P.; Taddei, M.L.; Paoli, P. miR-210-3p mediates metabolic adaptation and sustains DNA damage repair of resistant colon cancer cells to treatment with 5-fluorouracil. Mol. Carcinog. 2019, 58, 2181–2192. [Google Scholar] [CrossRef]
- Du, Y.; Wei, N.; Ma, R.; Jiang, S.; Song, D. A miR-210-3p regulon that controls the Warburg effect by modulating HIF-1α and p53 activity in triple-negative breast cancer. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Qiao, X.R.; Wang, L.; Liu, M.; Tian, Y.; Chen, T. MiR-210-3p attenuates lipid accumulation and inflammation in atherosclerosis by repressing IGF2. Biosci. Biotechnol. Biochem. 2020, 84, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Luo, R.; Liu, Y.; Gao, L.; Fu, Z.; Fu, Q.; Luo, X.; Chen, Y.; Deng, X.; Liang, Z.; et al. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR–p-PI3K/AKT-c-JUN. Nat. Commun. 2016, 7, 11309. [Google Scholar] [CrossRef] [PubMed]
- Pepe, F.; Pagotto, S.; Soliman, S.; Rossi, C.; Lanuti, P.; Braconi, C.; Mariani-Costantini, R.; Visone, R.; Veronese, A. Regulation of miR-483-3p by the O-linked N-acetylglucosamine transferase links chemosensitivity to glucose metabolism in liver cancer cells. Oncogenesis 2017, 6, 1–8. [Google Scholar] [CrossRef]
- Ji, H.; Yi, Q.; Chen, L.; Wong, L.; Liu, Y.; Xu, G.; Zhao, J.; Huang, T.; Li, B.; Yang, Y.; et al. Circulating miR-3197 and miR-2116-5p as novel biomarkers for diabetic retinopathy. Clin. Chim. Acta 2020, 501, 147–153. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Chen, Z.; Chen, J.; Meng, Y.; Chen, L.; Chang, Y.; Geng, B.; Sun, L.; Dou, L.; et al. NFE2 induces miR-423-5p to promote gluconeogenesis and hyperglycemia by repressing the hepatic FAM3A-ATP-Akt pathway. Diabetes 2017, 66, 1819–1832. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Zhang, Y.; Song, J. Potential proteins targeted by let-7f-5p in HeLa cells. Biosci. Trends 2017, 11, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Zhang, C.; Wei, X.; Wei, R.; Qi, Z.; Chen, K.; Cai, X.; Xu, L.; Tang, L.; Zhou, J.; et al. NEAT1/hsa-miR-372–3p axis participates in rapaMYCin-induced lipid metabolic disorder. Free Radic. Biol. Med. 2021, 167, 1–11. [Google Scholar] [CrossRef]
- Fierro-Fernández, M.; Miguel, V.; Márquez-Expósito, L.; Nuevo-Tapioles, C.; Herrero, J.I.; Blanco-Ruiz, E.; Tituaña, J.; Castillo, C.; Cannata, P.; Monsalve, M.; et al. MiR-9-5p protects from kidney fibrosis by metabolic reprogramming. FASEB J. 2020, 34, 410–431. [Google Scholar] [CrossRef]
- Wang, J.; Wang, B.; Ren, H.Q.; Chen, W. miR-9-5p inhibits pancreatic cancer cell proliferation, invasion and glutamine metabolism by targeting GOT1. Biochem. Biophys. Res. Commun. 2019, 509, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhu, X.; Yan, Z.; Li, C.; Zhao, H.; Ma, L.; Zhang, D.; Liu, J.; Liu, Z.; Du, N.; et al. miR-489-3p/SIX1 Axis Regulates Melanoma Proliferation and Glycolytic Potential. Mol. Ther. Oncolytics 2020, 16, 30–40. [Google Scholar] [CrossRef]
- Liu, N.; Feng, S.; Li, H.; Chen, X.; Bai, S.; Liu, Y. Long non-coding RNA MALAT1 facilitates the tumorigenesis, invasion and glycolysis of multiple myeloma via miR-1271-5p/SOX13 axis. J. Cancer Res. Clin. Oncol. 2020, 146, 367–379. [Google Scholar] [CrossRef]
- Woo, S.Y.; Lee, S.Y.; Yu, S.L.; Park, S.J.; Kang, D.; Kim, J.S.; Jeong, I.B.; Kwon, S.J.; Hwang, W.J.; Park, C.R.; et al. MicroRNA-7-5p′s role in the O-GlcNAcylation and cancer metabolism. Non-coding RNA Res. 2020, 5, 201–207. [Google Scholar] [CrossRef]
- Wang, F.; Fan, M.; Zhou, X.; Yu, Y.; Cai, Y.; Wu, H.; Zhang, Y.; Liu, J.; Huang, S.; He, N.; et al. A positive feedback loop between TAZ and miR-942-3p modulates proliferation, angiogenesis, epithelial-mesenchymal transition process, glycometabolism and ROS homeostasis in human bladder cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–19. [Google Scholar] [CrossRef]
- Deng, X.; Guo, B.; Fan, Y. Mir-153-3p suppresses cell proliferation, invasion and glycolysis of thyroid cancer through inhibiting e3f3 expression. OncoTargets Ther. 2021, 14, 519–529. [Google Scholar] [CrossRef]
- Wang, T.; Zhai, M.; Xu, S.; Ponnusamy, M.; Huang, Y.; Liu, C.Y.; Wang, M.; Shan, C.; Shan, P.P.; Gao, X.Q.; et al. NFATc3-dependent expression of miR-153-3p promotes mitochondrial fragmentation in cardiac hypertrophy by impairing mitofusin-1 expression. Theranostics 2020, 10, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Baresova, P.; Pitha, P.M.; Lubyova, B. Kaposi sarcoma-associated herpesvirus vIRF-3 protein binds to F-box of Skp2 protein and acts as a regulator of c-MYC protein function and stability. J. Biol. Chem. 2012, 287, 16199–16208. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Kellum, M.J.; Frisancho, J.A.; Pitha, P.M. Stimulation of c-MYC transcriptional activity by vIRF-3 of Kaposi sarcoma-associated herpesvirus. J. Biol. Chem. 2007, 282, 31944–31953. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-R.; Doganay, S.; Chung, B.; Toth, Z.; Brulois, K.; Lee, S.; Kanketayeva, Z.; Feng, P.; Ha, T.; Jung, J.U. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 4 (vIRF4) Targets Expression of Cellular IRF4 and the MYC Gene To Facilitate Lytic Replication. J. Virol. 2014, 88, 2183–2194. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Feng, J.; Deng, H.; Sun, R. MYC Is Required for the Maintenance of Kaposi’s Sarcoma-Associated Herpesvirus Latency. J. Virol. 2010, 84, 8945–8948. [Google Scholar] [CrossRef]
- Park, A.; Oh, S.; Jung, K.L.; Choi, U.Y.; Lee, H.R.; Rosenfeld, M.G.; Jung, J.U. Global epigenomic analysis of KSHV-infected primary effusion lymphoma identifies functional MYC superenhancers and enhancer RNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 21618–21627. [Google Scholar] [CrossRef]
- Rosean, T.R.; Holman, C.J.; Tompkins, V.S.; Jing, X.; Krasowski, M.D.; Rose-John, S.; Janz, S. KSHV-encoded vIL-6 collaborates with deregulated c-MYC to drive plasmablastic neoplasms in mice. Blood Cancer J. 2016, 6, e398. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Groshong, J.S.; Matta, H.; Schamus, S.; Punj, V.; Robinson, L.J.; Gill, P.S.; Chaudhary, P.M. Kaposi’s sarcoma associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 cooperates with MYC to promote lymphoma in mice. Cancer Biol. Ther. 2010, 10, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Carroll, P.A.; Thalhofer, A.B.; Lagunoff, M. Latent KSHV Infected Endothelial Cells Are Glutamine Addicted and Require Glutaminolysis for Survival. PLoS Pathog. 2015, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sarnow, P.; Ho, Y.S.; Williams, J.; Levine, A.J. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 1982, 28, 387–394. [Google Scholar] [CrossRef]
- Yew, P.R.; Berk, A.J. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 1992, 357, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.E.D.; Berk, A.J. Adenovirus E1B 55K Represses p53 Activation In Vitro. J. Virol. 1998, 72, 3146–3154. [Google Scholar] [CrossRef]
- Querido, E.; Morrison, M.R.; Chu-Pham-Dang, H.; Thirlwell, S.W.-L.; Boivin, D.; Branton, P.E. Identification of Three Functions of the Adenovirus E4orf6 Protein That Mediate p53 Degradation by the E4orf6-E1B55K Complex. J. Virol. 2001, 75, 2508. [Google Scholar] [CrossRef]
- Cathomen, T.; Weitzman, M.D. A Functional Complex of Adenovirus Proteins E1B-55kDa and E4orf6 Is Necessary To Modulate the Expression Level of p53 but Not Its Transcriptional Activity. J. Virol. 2000, 74, 11407–11412. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.M.; Kato, S.E.M.; Huang, W.; Flint, S.J.; Gonzalez, R.A. An early function of the adenoviral E1B 55 kDa protein is required for the nuclear relocalization of the cellular p53 protein in adenovirus-infected normal human cells. Virology 2008, 378, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Liao, D. Sequestration of p53 in the Cytoplasm by AdenovirusType 12 E1B 55-Kilodalton Oncoprotein Is Required for Inhibition ofp53-MediatedApoptosis. J. Virol. 2003, 77, 13171–13181. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Dobner, T. The adenovirus E1B-55K oncoprotein induces SUMO modification of p53. Cell Cycle 2008, 7, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Pennella, M.A.; Liu, Y.; Woo, J.L.; Kim, C.A.; Berk, A.J. Adenovirus E1B 55-Kilodalton Protein Is a p53-SUMO1 E3 Ligase That Represses p53 and Stimulates Its Nuclear Export through Interactions with Promyelocytic Leukemia Nuclear Bodies. J. Virol. 2010, 84, 12210–12225. [Google Scholar] [CrossRef]
- Lomonosova, E.; Subramanian, T.; Chinnadurai, G. Mitochondrial localization of p53 during adenovirus infection and regulation of its activity by E1B-19K. Oncogene 2005, 24, 6796–6808. [Google Scholar] [CrossRef][Green Version]
- Teodoro, J.G.; Shore, G.C.; Branton, P.E. Adenovirus E1A proteins induce apoptosis by both p53-dependent and p53-independent mechanisms. Oncogene 1995, 11, 467–474. [Google Scholar]
- Lowe, S.W.; Earl Ruley, H. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993, 7, 535–545. [Google Scholar] [CrossRef]
- Querido, E.; Teodoro, J.G.; Branton, P.E. Accumulation of p53 induced by the adenovirus E1A protein requires regions involved in the stimulation of DNA synthesis. J. Virol. 1997, 71, 3526–3533. [Google Scholar] [CrossRef]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. P53 Promotes Adenoviral Replication and Increases Late Viral Gene Expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef]
- Wright, J.; Leppard, K.N. The Human Adenovirus 5 L4 Promoter Is Activated by Cellular Stress Response Protein p53. J. Virol. 2013, 87, 11617–11625. [Google Scholar] [CrossRef][Green Version]
- Chahal, J.S.; Flint, S.J. The p53 Protein Does Not Facilitate Adenovirus Type 5 Replication in Normal Human Cells. J. Virol. 2013, 87, 6044–6046. [Google Scholar] [CrossRef]
- Steegenga, W.T.; van Laar, T.; Riteco, N.; Mandarino, A.; Shvarts, A.; van der Eb, A.J.; Jochemsen, A.G. Adenovirus E1A proteins inhibit activation of transcription by p53. Mol. Cell. Biol. 1996, 16, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Steegenga, W.T.; Shvarts, A.; Riteco, N.; Bos, J.L.; Jochemsen, A.G. Distinct Regulation of p53 and p73 Activity by Adenovirus E1A, E1B, and E4orf6 Proteins. Mol. Cell. Biol. 1999, 19, 3885–3894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frost, J.R.; Mendez, M.; Soriano, A.M.; Crisostomo, L.; Olanubi, O.; Radko, S.; Pelka, P. Adenovirus 5 E1A-Mediated Suppression of p53 via FUBP1. J. Virol. 2018, 92, e00439-18. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, N.; Usheva, A.; Chen, J.; Levine, A.J.; Weinmann, R.; Shenk, T. Two domains of p53 interact with the TATA-binding protein, and the adenovirus 13S E1A protein disrupts the association, relieving p53-mediated transcriptional repression. Mol. Cell. Biol. 1995, 15, 227–234. [Google Scholar] [CrossRef][Green Version]
- Lowe, S.W.; Jacks, T.; Housman, D.E.; Ruley, H.E. Abrogation of oncogene-associated apoptosis allows transformation of p53- deficient cells. Proc. Natl. Acad. Sci. USA 1994, 91, 2026–2030. [Google Scholar] [CrossRef] [PubMed]
- Nevels, M.; Spruss, T.; Wolf, H.; Dobner, T. The adenovirus E4orf6 protein contributes to malignant transformation by antagonizing E1A-induced accumulation of the tumor suppressor protein p53. Oncogene 1999, 18, 9–17. [Google Scholar] [CrossRef]
- Luo, K.; Ehrlich, E.; Xiao, Z.; Zhang, W.; Ketner, G.; Yu, X. Adenovirus E4orf6 assembles with Cullin5-ElonginB-ElonginC E3 ubiquitin ligase through an HIV/SIV Vif-like BC-box to regulate p53. FASEB J. 2007, 21, 1742–1750. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Shen, S.; Li, Y.; Bi, R.; Zhang, N.; Zheng, W.; Deng, Y.; Yang, Y.; Yu, X.F.; Wang, C.; et al. Adenovirus oncoprotein E4orf6 triggers Cullin5 neddylation to activate the CLR5 E3 ligase for p53 degradation. Biochem. Biophys. Res. Commun. 2019, 516, 1242–1247. [Google Scholar]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef]
- Blanchette, P.; Cheng, C.Y.; Yan, Q.; Ketner, G.; Ornelles, D.A.; Dobner, T.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Both BC-Box Motifs of Adenovirus Protein E4orf6 Are Required To Efficiently Assemble an E3 Ligase Complex That Degrades p53. Mol. Cell. Biol. 2004, 24, 9619–9629. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soria, C.; Estermann, F.E.; Espantman, K.C.; Oshea, C.C. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 2010, 466, 1076–1081. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; Zani, F.; Vousden, K.H. Control of metabolism by p53 – Cancer and beyond. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.S.; Mack, D.H.; Finicle, A.B.; Crook, T.; Vousden, K.H.; Laimins, L.A. Human papillomavirus E6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. EMBO J. 1992, 11, 3045–3052. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef]
- Li, S.; Hong, X.; Wei, Z.; Xie, M.; Li, W.; Liu, G.; Guo, H.; Yang, J.; Wei, W.; Zhang, S. Ubiquitination of the HPV oncoprotein E6 is critical for E6/E6AP-mediated p53 degradation. Front. Microbiol. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Conrady, M.C.; Suarez, I.; Gogl, G.; Frecot, D.I.; Bonhoure, A.; Kostmann, C.; Cousido-Siah, A.; Mitschler, A.; Lim, J.; Masson, M.; et al. Structure of High-Risk Papillomavirus 31 E6 Oncogenic Protein and Characterization of E6/E6AP/p53 Complex Formation. J. Virol. 2020, 95, e00730-20. [Google Scholar] [CrossRef]
- Yi, S.A.; Lee, D.H.; Kim, G.W.; Ryu, H.W.; Park, J.W.; Lee, J.; Han, J.; Park, J.H.; Oh, H.; Lee, J.; et al. HPV-mediated nuclear export of HP1γ drives cervical tumorigenesis by downregulation of p53. Cell Death Differ. 2020, 27, 2537–2551. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Thompson, D.A.; Münger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar]
- Massimi, P.; Banks, L. Repression of p53 transcriptional activity by the HPV E7 proteins. Virology 1997, 227, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Uxa, S.; Stanko, C.; Magin, T.M.; Engeland, K. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Prusinkiewicz, M.A.; Gameiro, S.F.; Ghasemi, F.; Dodge, M.J.; Zeng, P.Y.F.; Maekebay, H.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Survival-Associated Metabolic Genes in Human Papillomavirus-Positive Head and Neck Cancers. Cancers 2020, 12, 253. [Google Scholar] [CrossRef]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Jha, H.C.; El-Naccache, D.W.; Robertson, E.S. An EBV recombinant deleted for residues 130-159 in EBNA3C can deregulate p53/Mdm2 and Cyclin D1/CDK6 which results in apoptosis and reduced cell proliferation. Oncotarget 2016, 7, 18116–18134. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Bamidele, A.; Murakami, M.; Robertson, E.S. EBNA3C Attenuates the Function of p53 through Interaction with Inhibitor of Growth Family Proteins 4 and 5. J. Virol. 2011, 85, 2079–2088. [Google Scholar] [CrossRef]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-barr virus nuclear antigen 3C stabilizes gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011, 7, 1–12. [Google Scholar] [CrossRef]
- Shao, J.Y.; Ernberg, I.; Biberfeld, P.; Heiden, T.; Zeng, Y.X.; Hu, L.F. Epstein-Barr virus LMP1 status in relation to apoptosis, p53 expression and leucocyte infiltration in nasopharyngeal carcinoma. Anticancer Res. 2004, 24, 2309–2318. [Google Scholar]
- Fries, K.L.; Miller, W.E.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 1996, 70, 8653–8659. [Google Scholar] [CrossRef]
- Liu, M.T.; Chang, Y.T.; Chen, S.C.; Chuang, Y.C.; Chen, Y.R.; Lin, C.S.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 2005, 24, 2635–2646. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Chen, Y.; Jia, X.; Liu, Y. The anti-apoptotic role of EBV-LMP1 in lymphoma cells. Cancer Manag. Res. 2020, 12, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W.; Xiao, L.; Xu, J.; Chen, X.; Tang, M.; Dong, Z.; Tao, Q.; Cao, Y. Viral oncoprotein LMP1 disrupts p53-induced cell cycle arrest and apoptosis through modulating K63-linked ubiquitination of p53. Cell Cycle 2012, 11, 2327–2336. [Google Scholar] [CrossRef]
- Wang, Q.; Lingel, A.; Geiser, V.; Kwapnoski, Z.; Zhang, L. Tumor Suppressor p53 Stimulates the Expression of Epstein-Barr Virus Latent Membrane Protein 1. J. Virol. 2017, 91, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gutsch, D.; Kenney, S. Functional and physical interaction between p53 and BZLF1: Implications for Epstein-Barr virus latency. Mol. Cell. Biol. 1994, 14, 1929–1938. [Google Scholar] [CrossRef]
- Sato, Y.; Shirata, N.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Murata, T.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 2009, 388, 204–211. [Google Scholar] [CrossRef]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Li, L.; Liu, S.; Yang, L.; Ma, X.; Tang, M.; Bode, A.M.; Dong, Z.; Sun, L.; et al. EBV encoded miR-BHRF1-1 potentiates viral lytic replication by downregulating host p53 in nasopharyngeal carcinoma. Int. J. Biochem. Cell Biol. 2012, 44, 275–279. [Google Scholar] [CrossRef]
- Xu, D.M.; Kong, Y.L.; Wang, L.; Zhu, H.Y.; Wu, J.Z.; Xia, Y.; Li, Y.; Qin, S.C.; Fan, L.; Li, J.Y.; et al. EBV-mIR-BHRF1-1 targets p53 gene: Potential role in Epstein-Barr virus associated chronic lymphocytic leukemia. Cancer Res. Treat. 2020, 52, 492–504. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, J.; Wei, L.; Peng, Q.; Gao, Y.; Fu, Y.; Lu, Y.; Qin, Z.; Zhang, X.; Lu, J.; et al. Epstein-Barr Virus MicroRNA miR-BART5-3p Inhibits p53 Expression. J. Virol. 2018, 92, e01022-18. [Google Scholar] [CrossRef]
- Friborg, J.; Kong, W.P.; Hottlger, M.O.; Nabel, G.J. p53 Inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef]
- Cai, Q.L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006, 2, 1002–1012. [Google Scholar] [CrossRef]
- Suzuki, T.; Isobe, T.; Kitagawa, M.; Ueda, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA positively affects on ubiquitylation of p53. Biochem. Biophys. Res. Commun. 2010, 403, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Xiao, B.; Si, H.; Cervini, A.; Gao, J.; Lu, J.; Upadhyay, S.K.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 2012, 8, e1002566. [Google Scholar] [CrossRef]
- Si, H.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Latency-Associated Nuclear Antigen Induces Chromosomal Instability through Inhibition of p53 Function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef]
- Chen, W.; Hilton, I.B.; Staudt, M.R.; Burd, C.E.; Dittmer, D.P. Distinct p53, p53:LANA, and LANA Complexes in Kaposi’s Sarcoma-Associated Herpesvirus Lymphomas. J. Virol. 2010, 84, 3898–3908. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, D.; Kieffer-Kwon, P.; Ziegelbauer, J.M. Emerging themes from EBV and KSHV microRNA targets. Viruses 2012, 4, 1687–1710. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Li, M.; Zarycki, J.; Jung, J.U. Inhibition of p53 Tumor Suppressor by Viral Interferon Regulatory Factor. J. Virol. 2001, 75, 7572–7582. [Google Scholar] [CrossRef] [PubMed]
- Seo, T.; Park, J.; Lee, D.; Hwang, S.G.; Choe, J. Viral Interferon Regulatory Factor 1 of Kaposi’s Sarcoma-Associated Herpesvirus Binds to p53 and Represses p53-Dependent Transcription and Apoptosis. J. Virol. 2001, 75, 6193–6198. [Google Scholar] [CrossRef]
- Rivas, C.; Thlick, A.-E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus LANA2 Is a B-Cell-Specific Latent Viral Protein That Inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Baresova, P.; Musilova, J.; Pitha, P.M.; Lubyova, B. p53 Tumor Suppressor Protein Stability and Transcriptional Activity Are Targeted by Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Viral Interferon Regulatory Factor 3. Mol. Cell. Biol. 2014, 34, 386–399. [Google Scholar] [CrossRef]
- Laura, M.V.; De La Cruz-Herrera, C.F.; Ferreirós, A.; Baz-Martínez, M.; Lang, V.; Vidal, A.; Muñoz-Fontela, C.; Rodríguez, M.S.; Collado, M.; Rivas, C. KSHV latent protein LANA2 inhibits sumo2 modification of p53. Cell Cycle 2015, 14, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Villar, L.; Pérez-Girón, J.V.; Vilas, J.M.; Soto, A.; De La Cruz-Herrera, C.F.; Lang, V.; Collado, M.; Vidal, A.; Rodríguez, M.S.; Muñoz-Fontela, C.; et al. SUMOylation of p53 mediates interferon activities. Cell Cycle 2013, 12, 2809–2816. [Google Scholar] [CrossRef]
- Müller, S.; Berger, M.; Lehembre, F.; Seeler, J.S.; Haupt, Y.; Dejean, A. c-Jun and p53 activity is modulated by SUMO-1 modification. J. Biol. Chem. 2000, 275, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-R.; Toth, Z.; Shin, Y.C.; Lee, J.-S.; Chang, H.; Gu, W.; Oh, T.-K.; Kim, M.H.; Jung, J.U. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 4 Targets MDM2 To Deregulate the p53 Tumor Suppressor Pathway. J. Virol. 2009, 83, 6739–6747. [Google Scholar] [CrossRef] [PubMed]
- West, J.T.; Wood, C. The role of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 2003, 22, 5150–5163. [Google Scholar] [CrossRef]
- Gwack, Y.; Hwang, S.; Byun, H.; Lim, C.; Kim, J.W.; Choi, E.-J.; Choe, J. Kaposi’s Sarcoma-Associated Herpesvirus Open Reading Frame 50 Represses p53-Induced Transcriptional Activity and Apoptosis. J. Virol. 2001, 75, 6245–6248. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Konrad, A.; Jochmann, R.; Lausen, B.; Holz, P.; Naschberger, E.; Neipel, F.; Britzen-Laurent, N.; Stürzl, M. Structural proteins of Kaposi’s sarcoma-associated herpesvirus antagonize p53-mediated apoptosis. Oncogene 2015, 34, 639–649. [Google Scholar] [CrossRef]
- Alzhanova, D.; Corcoran, K.; Bailey, A.G.; Long, K.; Taft-Benz, S.; Graham, R.L.; Broussard, G.S.; Heise, M.; Neumann, G.; Halfmann, P.; et al. Novel modulators of p53-signaling encoded by unknown genes of emerging viruses. PLoS Pathog. 2021, 17, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-C.; Li, M. Kaposi’s Sarcoma-Associated Herpesvirus K-Cyclin Interacts with Cdk9 and Stimulates Cdk9-Mediated Phosphorylation of p53 Tumor Suppressor. J. Virol. 2008, 82, 278–290. [Google Scholar] [CrossRef]
- Balistreri, G.; Viiliäinen, J.; Turunen, M.; Diaz, R.; Lyly, L.; Pekkonen, P.; Rantala, J.; Ojala, K.; Sarek, G.; Teesalu, M.; et al. Oncogenic Herpesvirus Utilizes Stress-Induced Cell Cycle Checkpoints for Efficient Lytic Replication. PLoS Pathog. 2016, 12, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Lang, F.; Pei, Y.; Jha, H.C.; Robertson, E.S. Metabolic reprogramming of Kaposi’s sarcoma associated herpes virus infected B-cells in hypoxia. PLoS Pathog. 2018, 14, 1–28. [Google Scholar] [CrossRef]
- Choi, U.Y.; Lee, J.J.; Park, A.; Zhu, W.; Lee, H.R.; Choi, Y.J.; Yoo, J.S.; Yu, C.; Feng, P.; Gao, S.J.; et al. Oncogenic human herpesvirus hijacks proline metabolism for tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 8083–8093. [Google Scholar] [CrossRef] [PubMed]
- Moran, E. Interaction of adenoviral proteins with pRB and p53. FASEB J. 1993, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Whyte, P.; Buchkovich, K.J.; Horowitz, J.M.; Friend, S.H.; Raybuck, M.; Weinberg, R.A.; Harlow, E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988, 334, 124. [Google Scholar] [CrossRef]
- King, C.R.; Zhang, A.; Tessier, T.M.; Gameiro, S.F.; Mymryk, J.S. Hacking the Cell: Network Intrusion and Exploitation by Adenovirus E1A. MBio 2018, 9, e00390-18. [Google Scholar] [CrossRef]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor RB to repress select host genes and promote productive virus infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef]
- Ledl, A.; Schmidt, D.; Müller, S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene 2005, 24, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.Y.; Hearing, P. Adenovirus early proteins and host sumoylation. MBio 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.J.; Hearing, P. Mutually exclusive interaction of the adenovirus E4-6/7 protein and the retinoblastoma gene product with internal domains of E2F-1 and DP-1. J. Virol. 1994, 68, 6848–6862. [Google Scholar] [CrossRef]
- Schaley, J.E.; Polonskaia, M.; Hearing, P. The Adenovirus E4-6/7 Protein Directs Nuclear Localization of E2F-4 via an Arginine-Rich Motif. J. Virol. 2005, 79, 2301–2308. [Google Scholar] [CrossRef]
- Pelka, P.; Miller, M.S.; Cecchini, M.; Yousef, A.F.; Bowdish, D.M.; Dick, F.; Whyte, P.; Mymryk, J.S. Adenovirus E1A Directly Targets the E2F/DP-1 Complex. J. Virol. 2011, 85, 8841–8851. [Google Scholar] [CrossRef] [PubMed]
- Dallaire, F.; Schreiner, S.; Blair, G.E.; Dobner, T.; Branton, P.E.; Blanchette, P. The Human Adenovirus Type 5 E4orf6/E1B55K E3 Ubiquitin Ligase Complex Can Mimic E1A Effects on E2F. mSphere 2016, 1. [Google Scholar] [CrossRef]
- Dallaire, F.; Schreiner, S.; Blair, G.E.; Dobner, T.; Branton, P.E.; Blanchette, P. The Human Adenovirus Type 5 E4orf6/E1B55K E3 Ubiquitin Ligase Complex Enhances E1A Functional Activity. mSphere 2016, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.J.; Sage, J.; Duensing, S.; Münger, K.; Jacks, T.; Lambert, P.F. Recapitulation of the Effects of the Human Papillomavirus Type 16 E7 Oncogene on Mouse Epithelium by Somatic Rb Deletion and Detection of pRb-Independent Effects of E7 In Vivo. Mol. Cell. Biol. 2003, 23, 9094–9103. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.; Dick, F.; Lee, D.; Farrell, L.; Hyde, R.K.; Griep, A.E.; Dyson, N.; Lambert, P.F. Examination of the pRb-Dependent and pRb-Independent Functions of E7 In Vivo. J. Virol. 2005, 79, 11392–11402. [Google Scholar] [CrossRef]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adeno virus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Münger, K. Degradation of the Retinoblastoma Tumor Suppressor by the Human Papillomavirus Type 16 E7 Oncoprotein Is Important for Functional Inactivation and Is Separable from Proteasomal Degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [CrossRef]
- Collins, A.S.; Nakahara, T.; Do, A.; Lambert, P.F. Interactions with Pocket Proteins Contribute to the Role of Human Papillomavirus Type 16 E7 in the Papillomavirus Life Cycle. J. Virol. 2005, 79, 14769–14780. [Google Scholar] [CrossRef][Green Version]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef]
- Swenson, J.J.; Mauser, A.E.; Kaufmann, W.K.; Kenney, S.C. The Epstein-Barr Virus Protein BRLF1 Activates S Phase Entry through E2F1 Induction. J. Virol. 1999, 73, 6540–6550. [Google Scholar] [CrossRef]
- Zacny, V.L.; Wilson, J.; Pagano, J.S. The Epstein-Barr Virus Immediate-Early Gene Product, BRLF1, Interacts with the Retinoblastoma Protein during the Viral Lytic Cycle. J. Virol. 1998, 72, 8043–8051. [Google Scholar] [CrossRef] [PubMed]
- Parker, G.A.; Touitou, R.; Allday, M.J. Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene 2000, 19, 700–709. [Google Scholar] [CrossRef]
- Szekely, L.; Selivanova, G.; Magnusson, K.P.; Klein, G.; Wiman, K.G. EBNA-5, an Epstein-Barr virus-encoded nuclear antigen, binds to the retinoblastoma and p53 proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 5455–5459. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Lu, J.; Morizur, L.; Upadhyay, S.K.; AJ, M.P.; Robertson, E.S. E2F1 mediated apoptosis induced by the DNA damage response is blocked by EBV nuclear antigen 3C in lymphoblastoid cells. PLoS Pathog. 2012, 8, e1002573. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Banerjee, S.; Sun, Z.; Jha, H.C.; Saha, A.; Robertson, E.S. EBV Nuclear Antigen 3C Mediates Regulation of E2F6 to Inhibit E2F1 Transcription and Promote Cell Proliferation. PLoS Pathog. 2016, 12, 1–24. [Google Scholar] [CrossRef]
- Pei, Y.; Wong, J.H.; Jha, H.C.; Tian, T.; Wei, Z.; Robertson, E.S. Epstein-Barr Virus Facilitates Expression of KLF14 by Regulating the Cooperative Binding of the E2F-Rb-HDAC Complex in Latent Infection. J. Virol. 2020, 94, 1–16. [Google Scholar] [CrossRef]
- Lévy, P.; Bartosch, B. Metabolic reprogramming: A hallmark of viral oncogenesis. Oncogene 2016, 35, 4155–4164. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Kalejta, R.F. Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 2009, 4, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef]
- Godden-Kent, D.; Talbot, S.J.; Boshoff, C.; Chang, Y.; Moore, P.; Weiss, R.A.; Mittnacht, S. The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 1997, 71, 4193–4198. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Villar, L.; Gallego, P.; Muñoz-Fontela, C.; De La Cruz-Herrera, C.F.; Campagna, M.; González, D.; Lopitz-Otsoa, F.; Rodríguez, M.S.; Rivas, C. Kaposi’s sarcoma-associated herpesvirus lana2 protein interacts with the pocket proteins and inhibits their sumoylation. Oncogene 2014, 33, 495–503. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Cheng, F.; da Silva, S.R.; Tan, B.; Sorel, O.; Gruffaz, M.; Li, T.; Gao, S.-J. Molecular Biology of KSHV in Relation to HIV/AIDS-Associated Oncogenesis BT-HIV/AIDS-Associated Viral Oncogenesis. In HIV/AIDS-Associated Viral Oncogenesis; Meyers, C., Ed.; Springer International Publishing: Hershey, PA, USA, 2019; pp. 23–62. ISBN 978-3-030-03502-0. [Google Scholar]
- Cermelli, S.; Jang, I.S.; Bernard, B.; Grandori, C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb. Perspect. Med. 2014, 4, a014209. [Google Scholar] [CrossRef]
- Toyoshima, M.; Howie, H.L.; Imakura, M.; Walsh, R.M.; Annis, J.E.; Chang, A.N.; Frazier, J.; Chau, B.N.; Loboda, A.; Linsley, P.S.; et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Mast, F.D.; Navare, A.T.; van der Sloot, A.M.; Coulombe-Huntington, J.; Rout, M.P.; Baliga, N.S.; Kaushansky, A.; Chait, B.T.; Aderem, A.; Rice, C.M.; et al. Crippling life support for SARS-CoV-2 and other viruses through synthetic lethality. J. Cell Biol. 2020, 219, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dodge, M.J.; MacNeil, K.M.; Tessier, T.M.; Weinberg, J.B.; Mymryk, J.S. Emerging Antiviral Therapeutics for Human Adenovirus Infection: Recent Developments and Novel Strategies. Antiviral Res. 2021, 188, 105034. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prusinkiewicz, M.A.; Mymryk, J.S. Metabolic Control by DNA Tumor Virus-Encoded Proteins. Pathogens 2021, 10, 560. https://doi.org/10.3390/pathogens10050560
Prusinkiewicz MA, Mymryk JS. Metabolic Control by DNA Tumor Virus-Encoded Proteins. Pathogens. 2021; 10(5):560. https://doi.org/10.3390/pathogens10050560
Chicago/Turabian StylePrusinkiewicz, Martin A., and Joe S. Mymryk. 2021. "Metabolic Control by DNA Tumor Virus-Encoded Proteins" Pathogens 10, no. 5: 560. https://doi.org/10.3390/pathogens10050560
APA StylePrusinkiewicz, M. A., & Mymryk, J. S. (2021). Metabolic Control by DNA Tumor Virus-Encoded Proteins. Pathogens, 10(5), 560. https://doi.org/10.3390/pathogens10050560