
H3K4 Methylation in Aging and Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. H3K4 Methylation Marks Gene Expression across the Genome
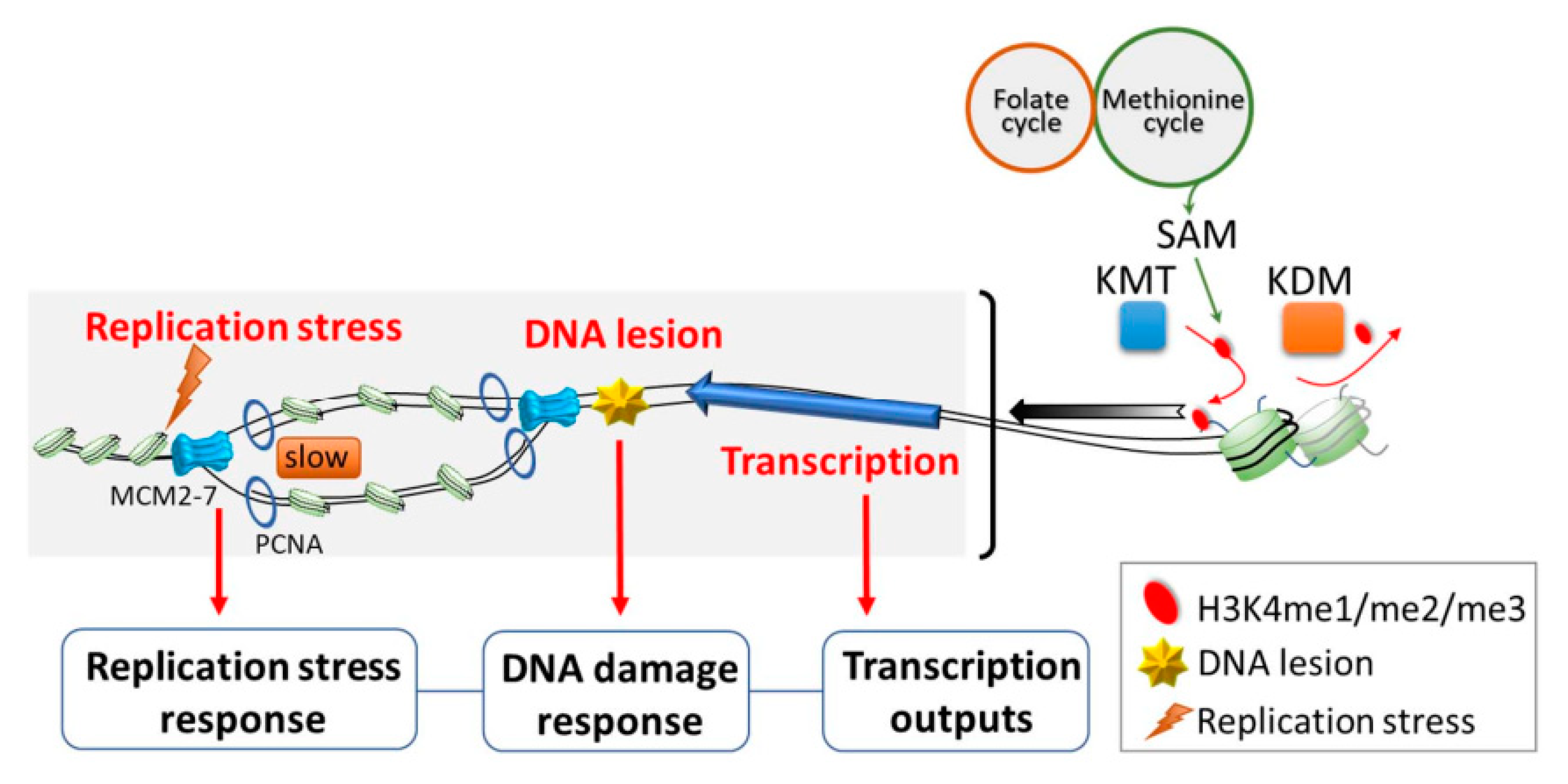
3. H3K4 Methylation Is Involved in Biological Functions Other than Transcription
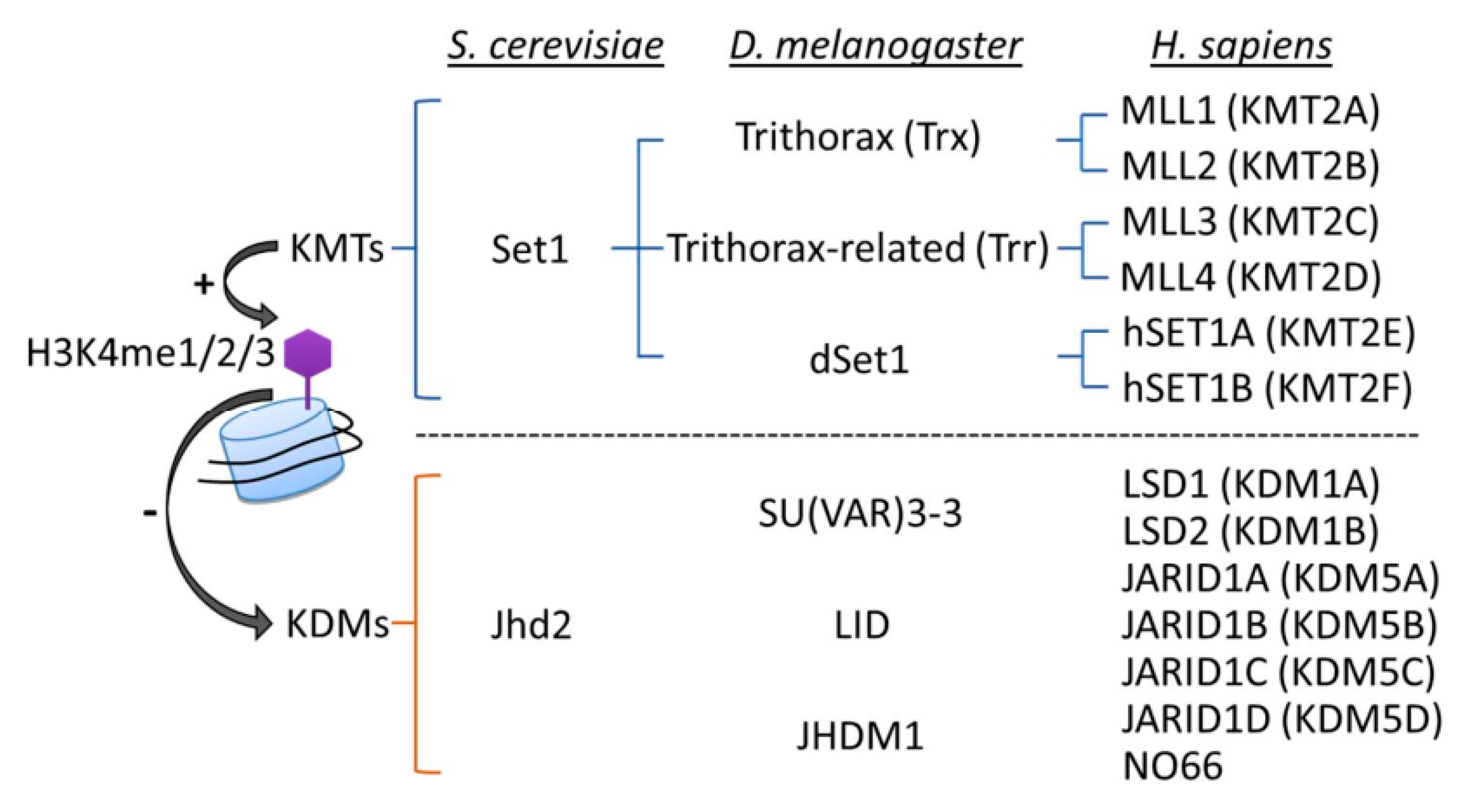
4. Writing and Erasing H3K4 Methylation
5. Roles of H3K4 Methylation in Regulating Aging in Different Species
5.1. H3K4 Methylation Contributes to Pathways That Regulate Yeast Aging and Lifespan
5.2. H3K4 Methylation Modulates Aging in Nematodes and Fruit Flies
5.3. H3K4 Methylation Is Associated with Age-Related Diseases in Mouse Models
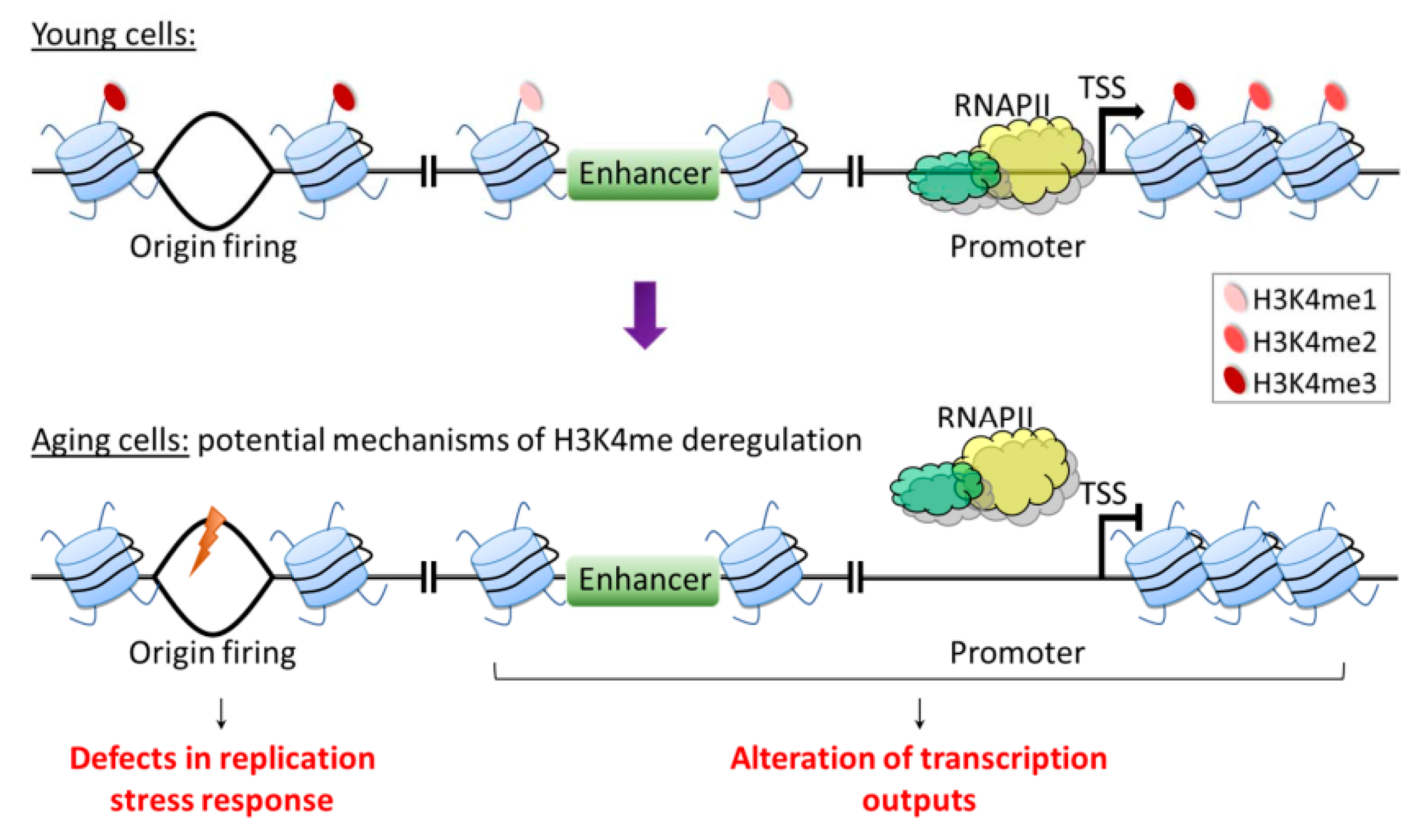
5.4. The Relationship between Regulation of H3K4 Methylation and Cell Senescence In Humans
6. Metabolic Signaling Pathways Involving H3K4 Methylation
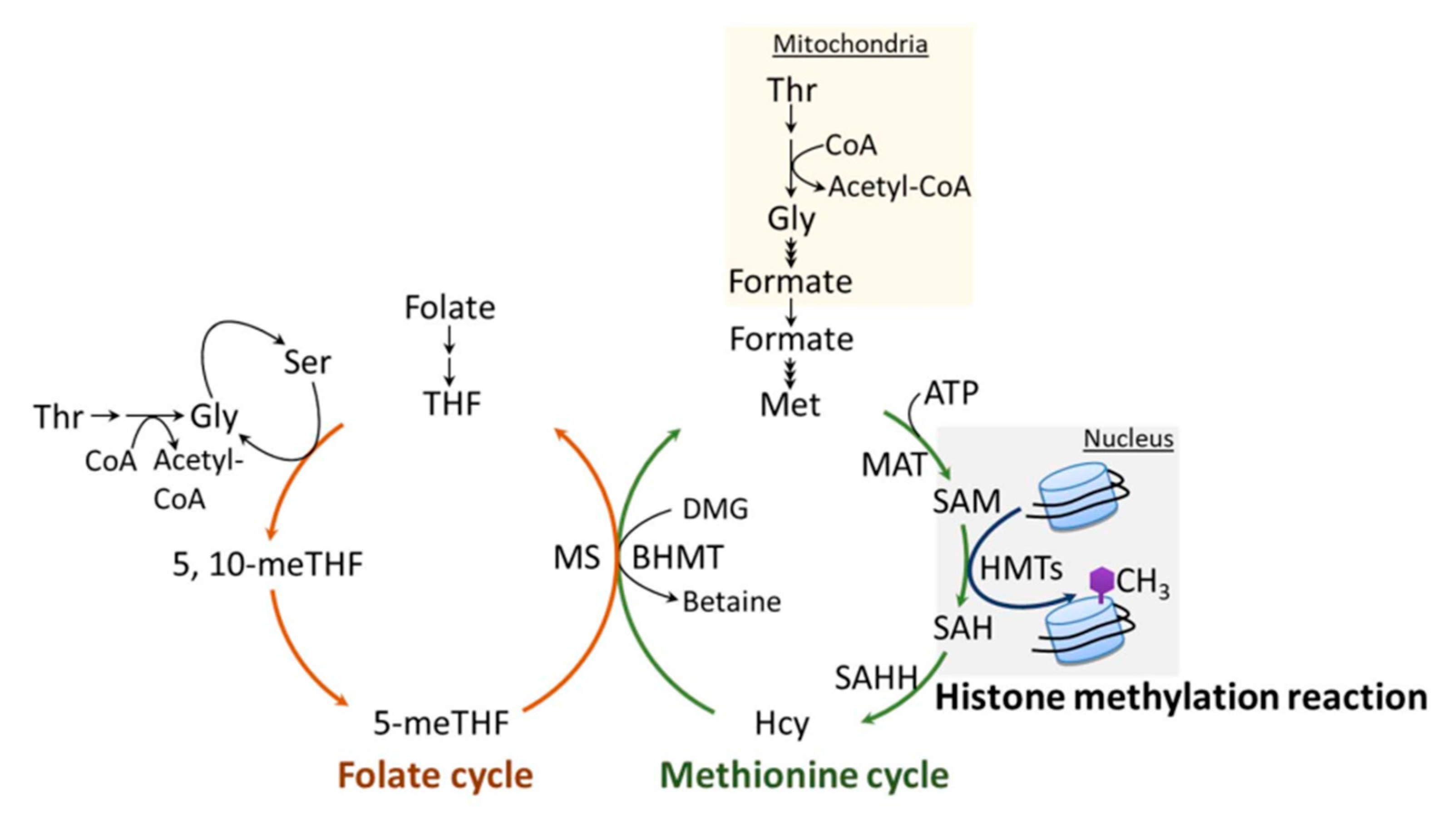
6.1. One-Carbon Metabolism
6.2. One-Carbon Metabolism and Its Regulation of H3K4 Methylation
6.3. Methionine and Its Involvement in Aging
7. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nat. Cell Biol. 2018, 561, 45–56. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending Healthy Life Span--From Yeast to Humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qu, J.; Liu, G.-H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Michalak, E.; Burr, M.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Gil, J. Cellular senescence causes ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 388. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Gensous, N.; Bacalini, M.G.; Pirazzini, C.; Marasco, E.; Giuliani, C.; Ravaioli, F.; Mengozzi, G.; Bertarelli, C.; Palmas, M.G.; Franceschi, C.; et al. The epigenetic landscape of age-related diseases: The geroscience perspective. Biogerontology 2017, 18, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Pagiatakis, C.; Musolino, E.; Gornati, R.; Bernardini, G.; Papait, R. Epigenetics of aging and disease: A brief overview. Aging Clin. Exp. Res. 2021, 33, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- de Magalhães, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signa-tures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.-H.; Chen, C.-C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Solis, G.M.; Thompson, R.C.; Gomez-Amaro, R.L.; Kurian, L.; Encalada, S.E.; Niculescu, A.B.; Salomon, D.R.; Petrascheck, M. Author response: Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. Author Response 2015, 4, 08833. [Google Scholar] [CrossRef]
- Frenk, S.; Houseley, J. Gene expression hallmarks of cellular ageing. Biogerontology 2018, 19, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Parkhitko, A.A.; Jouandin, P.; Mohr, S.E.; Perrimon, N. Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across spe-cies. Aging Cell 2019, 18, e13034. [Google Scholar] [CrossRef]
- Peleg, S.; Feller, C.; Ladurner, A.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef]
- Wood, I.C. The Contribution and Therapeutic Potential of Epigenetic Modifications in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kaya, A.; Ma, S.; Kim, G.; Gerashchenko, M.; Yim, S.H.; Hu, Z.; Harshman, L.G.; Gladyshev, V.N. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cabreiro, F.; Au, C.; Leung, K.-Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.; Gems, D. Metformin Retards Aging in C. elegans by Altering Microbial Folate and Methionine Metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Plummer, J.D.; Johnson, J.E. Extension of Cellular Lifespan by Methionine Restriction Involves Alterations in Central Carbon Metabolism and Is Mitophagy-Dependent. Front. Cell Dev. Biol. 2019, 7, 301. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Padilla, P.G.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Me-tabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine Metabolism Regulates Maintenance and Differentiation of Human Pluripotent Stem Cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nat. Cell Biol. 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nat. Cell Biol. 2015, 518, 413–416. [Google Scholar] [CrossRef]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, X.; Shen, J.; Wang, Y.; Li, X.; Liu, R.; Xia, Y.; Chen, Q.; Peng, G.; Lin, S.-Y.; et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 2015, 17, 1158–1168. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Im-munity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.; Braas, D.; Teitell, M.A. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485–493. [Google Scholar] [CrossRef]
- Ye, C.; Sutter, B.M.; Wang, Y.; Kuang, Z.; Tu, B.P. A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 2017, 66, 180–193.e8. [Google Scholar] [CrossRef]
- Maleszewska, M.; Mawer, J.S.P.; TessarzEmail, P. Histone Modifications in Ageing and Lifespan Regulation. Curr. Mol. Biol. Rep. 2016, 2, 26–35. [Google Scholar] [CrossRef]
- Molina-Serrano, D.; Kyriakou, D.; Kirmizis, A. Histone Modifications as an Intersection between Diet and Longevity. Front. Genet. 2019, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Mirsky, A.E.; Arnon, D.I.; Tsujimoto, H.Y.; McSwain, B.D. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science 1964, 144, 559. [Google Scholar] [CrossRef] [PubMed]
- Murray, K. The Occurrence of iε-N-Methyl Lysine in Histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Honda, B.M.; Dixon, G.H.; Candido, E.P. Sites of in vivo histone methylation in developing trout testis. J. Biol. Chem. 1975, 250, 8681–8685. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.; Sherriff, J.; Bernstein, B.E.; Emre, T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nat. Cell Biol. 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Leroy, G.; DiMaggio, P.A.; Chan, E.Y.; Zee, B.; Blanco, M.A.; Bryant, B.; Flaniken, I.Z.; Liu, S.; Kang, Y.; Trojer, P.; et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 2013, 6, 20. [Google Scholar] [CrossRef]
- Schibler, A.; Koutelou, E.; Tomida, J.; Wilson-Pham, M.; Wang, L.; Lu, Y.; Cabrera, A.P.; Chosed, R.J.; Li, W.; Li, B.; et al. Histone H3K4 methylation regulates deactivation of the spindle assembly checkpoint through direct binding of Mad2. Genes Dev. 2016, 30, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Beilharz, T.H.; Harrison, P.F.; Miles, D.M.; See, M.M.; Le, U.M.; Kalanon, M.; Curtis, M.J.; Hasan, Q.; Saksouk, J.; Margaritis, T.; et al. Coordination of Cell Cycle Progression and Mitotic Spindle Assembly Involves Histone H3 Lysine 4 Methyla-tion by Set1/COMPASS. Genetics 2017, 205, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Faucher, D.; Wellinger, R.J. Methylated H3K4, a Transcription-Associated Histone Modification, Is Involved in the DNA Damage Response Pathway. PLoS Genet. 2010, 6, e1001082. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Y.; Cutler, S.; Lin, J.-J.; Tsai, C.-H.; Tsai, H.-K.; Biggins, S.; Tsukiyama, T.; Lo, Y.-C.; Kao, C.-F. H3K4 methylation at active genes mitigates transcription-replication conflicts during replication stress. Nat. Commun. 2020, 11, 809. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Matter, A.; Fahrenkrog, B. Loss of histone H3 methylation at lysine 4 triggers apoptosis in Saccharomyces cere-visiae. PLoS Genet. 2014, 10, e1004095. [Google Scholar] [CrossRef] [PubMed]
- Rizzardi, L.F.; Dorn, E.S.; Strahl, B.D.; Cook, J.G. DNA Replication Origin Function Is Promoted by H3K4 Di-methylation in Saccharomyces cere-visiae. Genetics 2012, 192, 371. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Hu, H. H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 2014, 103, 222–228. [Google Scholar] [CrossRef]
- Pekowska, A.; Benoukraf, T.; Ferrier, P.; Spicuglia, S. A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res. 2010, 20, 1493–1502. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nat. Cell Biol. 2009, 459, 108–112. [Google Scholar] [CrossRef]
- Margaritis, T.; Oreal, V.; Brabers, N.; Maestroni, L.; Vitaliano-Prunier, A.; Benschop, J.J.; Van Hooff, S.; Van Leenen, D.; Dargemont, C.; Géli, V.; et al. Two Distinct Repressive Mechanisms for Histone 3 Lysine 4 Methylation through Promoting 3′-End Antisense Transcription. PLoS Genet. 2012, 8, e1002952. [Google Scholar] [CrossRef]
- Clouaire, T.; Webb, S.; Skene, P.; Illingworth, R.; Kerr, A.; Andrews, R.; Lee, J.-H.; Skalnik, D.; Bird, A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012, 26, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Howe, F.S.; Fischl, H.; Murray, S.C.; Mellor, J. Is H3K4me3 instructive for transcription activation? BioEssays 2017, 39, e201600095-12. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3K4 Methylation Patterns. Mol. Cell 2017, 68, 773–785.e6. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.; Devarajan, K.; Daugherty, A.C.; Kundaje, A.; Mancini, E.; et al. H3K4me3 Breadth Is Linked to Cell Identity and Transcriptional Consistency. Cell 2014, 158, 673–688. [Google Scholar] [CrossRef]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015, 47, 1149–1157. [Google Scholar] [CrossRef]
- Dincer, A.; Gavin, D.P.; Xu, K.; Zhang, B.; Dudley, J.T.; Schadt, E.E.; Akbarian, S. Deciphering H3K4me3 broad domains associated with gene-regulatory networks and conserved epigenomic land-scapes in the human brain. Transl. Psychiatry 2015, 5, e679. [Google Scholar] [CrossRef]
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. eLife 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Pu, M.; Wang, M.; Wang, W.; Velayudhan, S.S.; Lee, S.S. Unique patterns of trimethylation of histone H3 lysine 4 are prone to changes during aging in Caenorhabditis elegans somatic cells. PLoS Genet. 2018, 14, e1007466. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, D.; Jezek, M.; Quijote, J.; Lum, J.; Choi, G.; Kulkarni, R.; Park, D.; Green, E.M. Repression of Middle Sporulation Genes in Saccharomyces cerevisiae by the Sum1-Rfm1-Hst1 Complex Is Main-tained by Set1 and H3K4 Methylation. G3 Genes Genomes Genet. 2017, 7, 3971–3982. [Google Scholar]
- Karnani, N.; Taylor, C.M.; Malhotra, A.; Dutta, A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell 2010, 21, 393–404. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Synder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Karnani, N.; Taylor, C.; Malhotra, A.; Dutta, A. Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res. 2007, 17, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.P.; Labib, K. Chromosome Duplication in Saccharomyces cerevisiae. Genet. 2016, 203, 1027–1067. [Google Scholar] [CrossRef] [PubMed]
- MacAlpine, D.M.; Almouzni, G. Chromatin and DNA Replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010207. [Google Scholar] [CrossRef] [PubMed]
- Bleichert, F.; Botchan, M.R.; Berger, J.M. Mechanisms for initiating cellular DNA replication. Science 2017, 355, eaah6317. [Google Scholar] [CrossRef] [PubMed]
- Labib, K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010, 24, 1208–1219. [Google Scholar] [CrossRef]
- Natsume, T.; Müller, C.A.; Katou, Y.; Retkute, R.; Gierliński, M.; Araki, H.; Blow, J.J.; Shirahige, K.; Nieduszynski, C.A.; Tanaka, T.U. Kinetochores Coordinate Pericentromeric Cohesion and Early DNA Replication by Cdc7-Dbf4 Kinase Recruit-ment. Mol. Cell 2013, 50, 661–674. [Google Scholar] [CrossRef]
- Fang, D.; Lengronne, A.; Shi, D.; Forey, R.; Skrzypczak, M.; Ginalski, K.; Yan, C.; Wang, X.; Cao, Q.; Pasero, P.; et al. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. 2017, 31, 2405–2415. [Google Scholar] [CrossRef]
- Knott, S.R.; Peace, J.M.; Ostrow, A.Z.; Gan, Y.; Rex, A.E.; Viggiani, C.J.; Tavaré, S.; Aparicio, O.M. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell 2012, 148, 99–111. [Google Scholar] [CrossRef]
- Rhind, N.R.; Gilbert, D.M. DNA Replication Timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef]
- Pohl, T.J.; Brewer, B.J.; Raghuraman, M.K. Functional Centromeres Determine the Activation Time of Pericentric Origins of DNA Replication in Saccharomyces cerevisiae. PLoS Genet. 2012, 8, e1002677. [Google Scholar] [CrossRef] [PubMed]
- Vogelauer, M.; Rubbi, L.; Lucas, I.; Brewer, B.J.; Grunstein, M. Histone acetylation regulates the time of replication origin firing. Molecular Cell 2002, 10, 1223–1233. [Google Scholar] [CrossRef]
- Radman-Livaja, M.; Liu, C.L.; Friedman, N.; Schreiber, S.L.; Rando, O.J. Replication and Active Demethylation Represent Partially Overlapping Mechanisms for Erasure of H3K4me3 in Budding Yeast. PLoS Genet. 2010, 6, e1000837. [Google Scholar] [CrossRef]
- Rampakakis, E.; Di Paola, D.; Chan, M.K.; Zannis-Hadjopoulos, M. Dynamic changes in chromatin structure through post-translational modifications of histone H3 during rep-lication origin activation. J. Cell. Biochem. 2009, 108, 400–407. [Google Scholar] [CrossRef]
- Rondinelli, B.; Schwerer, H.; Antonini, E.; Gaviraghi, M.; Lupi, A.; Frenquelli, M.; Cittaro, D.; Segalla, S.; Lemaitre, J.-M.; Tonon, G. H3K4me3 demethylation by the histone demethylase KDM5C/JARID1C promotes DNA replication origin firing. Nucleic Acids Res. 2015, 43, 2560–2574. [Google Scholar] [CrossRef]
- Higgs, M.R.; Sato, K.; Reynolds, J.J.; Begum, S.; Bayley, R.; Goula, A.; Vernet, A.; Paquin, K.L.; Skalnik, D.G.; Kobayashi, W.; et al. Histone Methylation by SETD1A Protects Nascent DNA through the Nucleosome Chaperone Activity of FANCD2. Mol. Cell 2018, 71, 25–41.e6. [Google Scholar] [CrossRef]
- Shilatifard, A. The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Path-ogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Mohan, M.; Herz, H.M.; Smith, E.R.; Zhang, Y.; Jackson, J.; Washburn, M.P.; Florens, L.; Eissenberg, J.C.; Shilatifard, A. The COMPASS family of H3K4 methylases in Drosophila. Mol. Cell Biol. 2011, 31, 4310–4318. [Google Scholar] [CrossRef]
- Ardehali, M.B.; Mei, A.; Zobeck, K.L.; Caron, M.; Lis, J.T.; Kusch, T. DrosophilaSet1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. Embo. J. 2011, 30, 2817–2828. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Qu, Q.; Takahashi, Y.-H.; Yang, Y.; Hu, H.; Zhang, Y.; Brunzelle, J.S.; Couture, J.-F.; Shilatifard, A.; Skiniotis, G. Structure and Conformational Dynamics of a COMPASS Histone H3K4 Methyltransferase Complex. Cell 2018, 174, 1117–1126.e12. [Google Scholar] [CrossRef]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A novel non-SET domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methyla-tion by the mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2011, 286, 3359–3369. [Google Scholar] [CrossRef]
- Li, T.; Kelly, W.G. A Role for Set1/MLL-Related Components in Epigenetic Regulation of the Caenorhabditis elegans Germ Line. PLoS Genet. 2011, 7, e1001349. [Google Scholar] [CrossRef]
- Xiao, Y.; Bedet, C.; Robert, V.J.P.; Simonet, T.; Dunkelbarger, S.; Rakotomalala, C.; Soete, G.; Korswagen, H.C.; Strome, S.; Palladino, F. Caenorhabditis elegans chromatin-associated proteins SET-2 and ASH-2 are differentially required for histone H3 Lys 4 methylation in embryos and adult germ cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8305–8310. [Google Scholar] [CrossRef]
- Lefevre, G.M.; Patel, S.R.; Kim, D.; Tessarollo, L.; Dressler, G.R. Altering a Histone H3K4 Methylation Pathway in Glomerular Podocytes Promotes a Chronic Disease Pheno-type. PLoS Genet. 2010, 6, e1001142. [Google Scholar] [CrossRef]
- Lee, J.H.; Skalnik, D.G. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltrans-ferase complex to transcription start sites of transcribed human genes. Mol. Cell Biol. 2008, 28, 609–618. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Jothi, R. Genome-Wide Characterization of Menin-Dependent H3K4me3 Reveals a Specific Role for Menin in the Regulation of Genes Implicated in MEN1-Like Tumors. PLoS ONE 2012, 7, e37952. [Google Scholar] [CrossRef]
- Byvoet, P.; Shepherd, G.; Hardin, J.; Noland, B. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 1972, 148, 558–567. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Hojfeldt, J.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef]
- Sinha, K.M.; Yasuda, H.; Coombes, M.M.; Dent, S.; De Crombrugghe, B. Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. Embo. J. 2009, 29, 68–79. [Google Scholar] [CrossRef]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef]
- Christensen, J.; Agger, K.; Cloos, P.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. RBP2 Belongs to a Family of Demethylases, Specific for Tri-and Dimethylated Lysine 4 on Histone 3. Cell 2007, 128, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Nottke, A.C.; Beese-Sims, S.E.; Pantalena, L.F.; Reinke, V.; Shi, Y.; Colaiácovo, M.P. SPR-5 is a histone H3K4 demethylase with a role in meiotic double-strand break repair. Proc. Natl. Acad. Sci. USA 2011, 108, 12805–12810. [Google Scholar] [CrossRef]
- Greer, E.L.; Becker, B.; Latza, C.; Antebi, A.; Shi, Y. Mutation of C. elegans demethylase spr-5 extends transgenerational longevity. Cell Res. 2015, 26, 229–238. [Google Scholar] [CrossRef]
- Alvares, S.M.; Mayberry, G.A.; Joyner, E.Y.; Lakowski, B.; Ahmed, S. H3K4 demethylase activities repress proliferative and postmitotic aging. Aging Cell 2013, 13, 245–253. [Google Scholar] [CrossRef]
- Wang, S.; Meyer, D.H.; Schumacher, B. H3K4me2 regulates the recovery of protein biosynthesis and homeostasis following DNA damage. Nat. Struct. Mol. Biol. 2020, 27, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.J.; Edwards, T.M.; Reinke, V.; Kelly, W.G. A C. elegans LSD1 Demethylase Contributes to Germline Immortality by Reprogramming Epigenetic Memory. Cell 2009, 137, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.; Maures, T.J.; Hauswirth, A.G.; Green, E.M.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nat. Cell Biol. 2010, 466, 383–387. [Google Scholar] [CrossRef]
- Maures, T.J.; Greer, E.; Hauswirth, A.G.; Brunet, A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 2011, 10, 980–990. [Google Scholar] [CrossRef]
- Kidder, B.L.; Hu, G.; Zhao, K. KDM5B focuses H3K4 methylation near promoters and enhancers during embryonic stem cell self-renewal and differentiation. Genome Biol. 2014, 15, R32. [Google Scholar] [CrossRef]
- Outchkourov, N.S.; Muiño, J.M.; Kaufmann, K.; van IJcken, W.F.; Koerkamp, M.J.; van Leenen, D.; de Graaf, P.; Holstege, F.C.; Grosveld, F.G.; Timmers, H.M. Balancing of Histone H3K4 Methylation States by the Kdm5c/SMCX Histone Demethylase Mod-ulates Promoter and Enhancer Function. Cell Rep. 2013, 3, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Lussi, Y.; Mariani, L.; Friis, C.; Peltonen, J.; Myers, T.R.; Krag, C.; Wong, G.; Salcini, A.E. Impaired removal of H3K4 methylation affects cell fate determination and gene transcription. Development 2016, 143, 3751–3762. [Google Scholar] [CrossRef]
- Longo, V.D.; Fabrizio, P. Chronological Aging in Saccharomyces cerevisiae. Prokaryotic Cytoskelet. 2011, 57, 101–121. [Google Scholar] [CrossRef]
- Sinclair, D. Studying the replicative life span of yeast cells. Breast Cancer 2013, 1048, 49–63. [Google Scholar] [CrossRef]
- Lee, S.S.; Kennedy, S.; Tolonen, A.C.; Ruvkun, G. DAF-16 Target Genes That Control, C. elegans Life-Span and Metabolism. Science 2003, 300, 644–647. [Google Scholar] [CrossRef]
- Ni, Z.; Ebata, A.; Alipanahiramandi, E.; Lee, S.S. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 2011, 11, 315–325. [Google Scholar] [CrossRef] [PubMed]
- McColl, G.; Killilea, D.W.; Hubbard, A.E.; Vantipalli, M.C.; Melov, S.; Lithgow, G.J. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J. Biol. Chem. 2008, 283, 350–357. [Google Scholar] [CrossRef]
- Li, L.; Greer, C.; Eisenman, R.N.; Secombe, J. Essential Functions of the Histone Demethylase Lid. PLoS Genet. 2010, 6, e1001221. [Google Scholar] [CrossRef] [PubMed]
- Siebold, A.P.; Banerjee, R.; Tie, F.; Kiss, D.; Moskowitz, J.; Harte, P.J. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Adelman, E.R.; Huang, H.-T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, W.; Williams, J.B.; Yang, F.; Wang, Z.-J.; Yan, Z. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci. Adv. 2020, 6, eabc8096. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, M.B.; Jha, P.; Houtkooper, R.; Williams, R.W.; Wang, X.; Auwerx, J. The Gene-Regulatory Footprint of Aging Highlights Conserved Central Regulators. Cell Rep. 2020, 32, 108203. [Google Scholar] [CrossRef]
- Shafiee, G.; Keshtkar, A.; Soltani, A.; Ahadi, Z.; Larijani, B.; Heshmat, R. Prevalence of sarcopenia in the world: A systematic review and meta- analysis of general population studies. J. Diabetes Metab. Disord. 2017, 16, 1–10. [Google Scholar] [CrossRef]
- Sayer, A.A.; Cooper, C. Fetal programming of body composition and musculoskeletal development. Early Hum. Dev. 2005, 81, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.P.; Al-Shanti, N.; Davies, L.C.; Barton, S.J.; Grounds, M.D.; Tellam, R.L.; Stewart, C.E.; Cooper, C.; Sayer, A.A. Lean mass, muscle strength and gene expression in community dwelling older men: Findings from the Hertford-shire Sarcopenia Study (HSS). Calcif. Tissue Int. 2014, 95, 308–316. [Google Scholar] [CrossRef]
- Sharples, A.P.; Stewart, C.E.; Seaborne, R.A. Does skeletal muscle have an ‘epi’-memory? The role of epigenetics in nutritional programming, metabolic disease, aging and exercise. Aging Cell 2016, 15, 603–616. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Ong, C.-T.; Corces, V.G. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef]
- Hsieh, T.-H.S.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef]
- Weiner, A.; Hsieh, T.-H.S.; Appleboim, A.; Chen, H.V.; Rahat, A.; Amit, I.; Rando, O.J.; Friedman, N. High-Resolution Chromatin Dynamics during a Yeast Stress Response. Mol. Cell 2015, 58, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, aaf1420. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, G. S-adenosylmethionine. A new intermediate formed enzymatically from l-methionine and adenosinetriphosphate. J. Biol. Chem. 1953, 204, 403–416. [Google Scholar] [CrossRef]
- Teperino, R.; Schoonjans, K.; Auwerx, J. Histone Methyl Transferases and Demethylases; Can They Link Metabolism and Transcription? Cell Metab. 2010, 12, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Tehlivets, O.; Malanovic, N.; Visram, M.; Pavkov-Keller, T.; Keller, W. S-adenosyl-L-homocysteine hydrolase and methylation disorders: Yeast as a model system. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.-C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nat. Cell Biol. 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic meth-ylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J.; et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of me-thyl-deficient cystathionine beta-synthase heterozygous mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, M.; Guan, Q.; Li, F.; Sales-Lee, J.; Iavarone, A.T.; Hammond, M.C.; Cande, W.Z.; Rine, J. Nutritional Control of Epigenetic Processes in Yeast and Human Cells. Genetics 2013, 195, 831–844. [Google Scholar] [CrossRef]
- Dai, Z.; Mentch, S.J.; Gao, X.; Nichenametla, S.N.; Locasale, J.W. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Kera, Y.; Katoh, Y.; Ohta, M.; Matsumoto, M.; Takano-Yamamoto, T.; Igarashi, K. Methionine Adenosyltransferase II-dependent Histone H3K9 Methylation at the COX-2 Gene Locus. J. Biol. Chem. 2013, 288, 13592–13601. [Google Scholar] [CrossRef]
- Hayashi, T.; Teruya, T.; Chaleckis, R.; Morigasaki, S.; Yanagida, M. S-Adenosylmethionine Synthetase Is Required for Cell Growth, Maintenance of G0 Phase, and Termination of Quiescence in Fission Yeast. iScience 2018, 5, 38–51. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Miller, J.W.; Da Costa, K.-A.; Nadeau, M.; Smith, D.; Selhub, J.; Zeisel, S.H.; Mason, J.B. Severe Folate Deficiency Causes Secondary Depletion of Choline and Phosphocholine in Rat Liver. J. Nutr. 1994, 124, 2197–2203. [Google Scholar] [CrossRef]
- Miller, J.W.; Nadeau, M.R.; Smith, J.; Smith, D.; Selhub, J. Folate-deficiency-induced homocysteinaemia in rats: Disruption of S-adenosylmethionine’s co-ordinate regulation of homocysteine metabolism. Biochem. J. 1994, 298, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Balaghi, M.; Horne, D.W.; Wagner, C. Hepatic one-carbon metabolism in early folate deficiency in rats. Biochem. J. 1993, 291, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Pogribny, I.P.; Basnakian, A.G.; Miller, J.W.; Selhub, J.; James, S.J.; Mason, J.B. Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumor suppressor gene. Am. J. Clin. Nutr. 1997, 65, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Stempak, J.M.; Sohn, K.-J.; Chiang, E.-P.; Shane, B.; Kim, Y.-I. Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model. Carcinogenesis 2005, 26, 981–990. [Google Scholar] [CrossRef]
- Alonso-Aperte, E.; González, M.P.; Póo-Prieto, R.; Varela-Moreiras, G. Folate status and S-adenosylmethionine/S-adenosylhomocysteine ratio in colorectal adenocarcinoma in humans. Eur. J. Clin. Nutr. 2007, 62, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Rivera, I.; Struys, E.A.; Jansen, E.E.; Ravasco, P.; Camilo, M.E.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular dis-ease. Clin. Chem. 2003, 49, 1292–1296. [Google Scholar] [CrossRef]
- Luka, Z.; Moss, F.; Loukachevitch, L.V.; Bornhop, D.J.; Wagner, C. Histone Demethylase LSD1 Is a Folate-Binding Protein. Biochemistry 2011, 50, 4750–4756. [Google Scholar] [CrossRef] [PubMed]
- Luka, Z.; Pakhomova, S.; Loukachevitch, L.V.; Calcutt, M.W.; Newcomer, M.E.; Wagner, C. Crystal structure of the histone lysine specific demethylase LSD1 complexed with tetrahydrofolate. Protein Sci. 2014, 23, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Luka, Z.; Loukachevitch, L.V.; Bhanu, N.V.; Wagner, C. Folate deficiency affects histone methylation. Med Hypotheses 2016, 88, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ruckenstuhl, C.; Netzberger, C.; Entfellner, I.; Carmona-Gutierrez, D.; Kickenweiz, T.; Stekovic, S.; Gleixner, C.; Schmid, C.; Klug, L.; Sorgo, A.G.; et al. Lifespan Extension by Methionine Restriction Requires Autophagy-Dependent Vacuolar Acidification. PLoS Genet. 2014, 10, e1004347. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef]
- Bárcena, C.; Quiros, P.M.; Durand, S.; Mayoral, P.; Rodríguez, F.; Caravia, X.M.; Mariño, G.; Garabaya, C.; Fernández-García, M.T.; Kroemer, G.; et al. Methionine Restriction Extends Lifespan in Progeroid Mice and Alters Lipid and Bile Acid Metabolism. Cell Rep. 2018, 24, 2392–2403. [Google Scholar] [CrossRef]
- Sharma, S.; Dixon, T.; Jung, S.; Graff, E.C.; Forney, L.A.; Gettys, T.W.; Wanders, D. Dietary Methionine Restriction Reduces Inflammation Independent of FGF21 Action. Obesity 2019, 27, 1305–1313. [Google Scholar] [CrossRef]
- Maddineni, S.; Nichenametla, S.; Sinha, R.; Wilson, R.P.; Richie, J.P. Methionine restriction affects oxidative stress and glutathione-related redox pathways in the rat. Exp. Biol. Med. 2013, 238, 392–399. [Google Scholar] [CrossRef]
- Ying, Y.; Yun, J.; Guoyao, W.; Kaiji, S.; Zhaolai, D.; Zhenlong, W. Dietary l-methionine restriction decreases oxidative stress in porcine liver mitochondria. Exp. Gerontol. 2015, 65, 35–41. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Sun, J.; Zhang, J.; Guo, H.; Shi, Y.-H.; Cheng, X.; Tang, X.; Le, G. Dietary methionine restriction reduces hepatic steatosis and oxidative stress in high-fat-fed mice by promoting H2S production. Food Funct. 2018, 10, 61–77. [Google Scholar] [CrossRef]
- Orgeron, M.L.; Stone, K.P.; Wanders, D.; Cortez, C.C.; Van, N.T.; Gettys, T.W. The Impact of Dietary Methionine Restriction on Biomarkers of Metabolic Health. Prog. Mol. Biol. Transl. Sci. 2014, 121, 351–376. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Greenway, F.L.; Boudreau, A.; Hill, K.L.; Johnson, W.; Krajcik, R.A.; Perrone, C.E.; Orentreich, N.; Cefalu, W.T.; Gettys, T.W. Dietary Methionine Restriction Increases Fat Oxidation in Obese Adults with Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E836–E840. [Google Scholar] [CrossRef]
- Wanders, D.; Forney, L.A.; Stone, K.P.; Burk, D.H.; Pierse, A.; Gettys, T.W. FGF21 Mediates the Thermogenic and Insulin-Sensitizing Effects of Dietary Methionine Restriction but Not Its Effects on Hepatic Lipid Metabolism. Diabetes 2017, 66, 858–867. [Google Scholar] [CrossRef]
- Ables, G.P.; Perrone, C.E.; Orentreich, D.; Orentreich, N. Methionine-Restricted C57BL/6J Mice Are Resistant to Diet-Induced Obesity and Insulin Resistance but Have Low Bone Density. PLoS ONE 2012, 7, e51357. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Johnson, F.B. Methionine Restriction Activates the Retrograde Response and Confers Both Stress Tolerance and Lifespan Extension to Yeast, Mouse and Human Cells. PLoS ONE 2014, 9, e97729. [Google Scholar] [CrossRef]
- Mattocks, D.A.; Mentch, S.J.; Shneyder, J.; Ables, G.P.; Sun, D.; Richie, J.P.; Locasale, J.W.; Nichenametla, S.N. Short term methionine restriction increases hepatic global DNA methylation in adult but not young male C57BL/6J mice. Exp. Gerontol. 2017, 88, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 2020, 31, 250–266.e9. [Google Scholar] [CrossRef]
- Ioannidou, A.; Goulielmaki, E.; Garinis, G.A. DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front. Genet. 2016, 7, 187. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage—How and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-L.; Lo, Y.-C.; Kao, C.-F. H3K4 Methylation in Aging and Metabolism. Epigenomes 2021, 5, 14. https://doi.org/10.3390/epigenomes5020014
Hsu C-L, Lo Y-C, Kao C-F. H3K4 Methylation in Aging and Metabolism. Epigenomes. 2021; 5(2):14. https://doi.org/10.3390/epigenomes5020014
Chicago/Turabian StyleHsu, Chia-Ling, Yi-Chen Lo, and Cheng-Fu Kao. 2021. "H3K4 Methylation in Aging and Metabolism" Epigenomes 5, no. 2: 14. https://doi.org/10.3390/epigenomes5020014
APA StyleHsu, C.-L., Lo, Y.-C., & Kao, C.-F. (2021). H3K4 Methylation in Aging and Metabolism. Epigenomes, 5(2), 14. https://doi.org/10.3390/epigenomes5020014