
The Contribution of Epigenetic Inheritance Processes on Age-Related Cognitive Decline and Alzheimer’s Disease




Abstract
1. Introduction
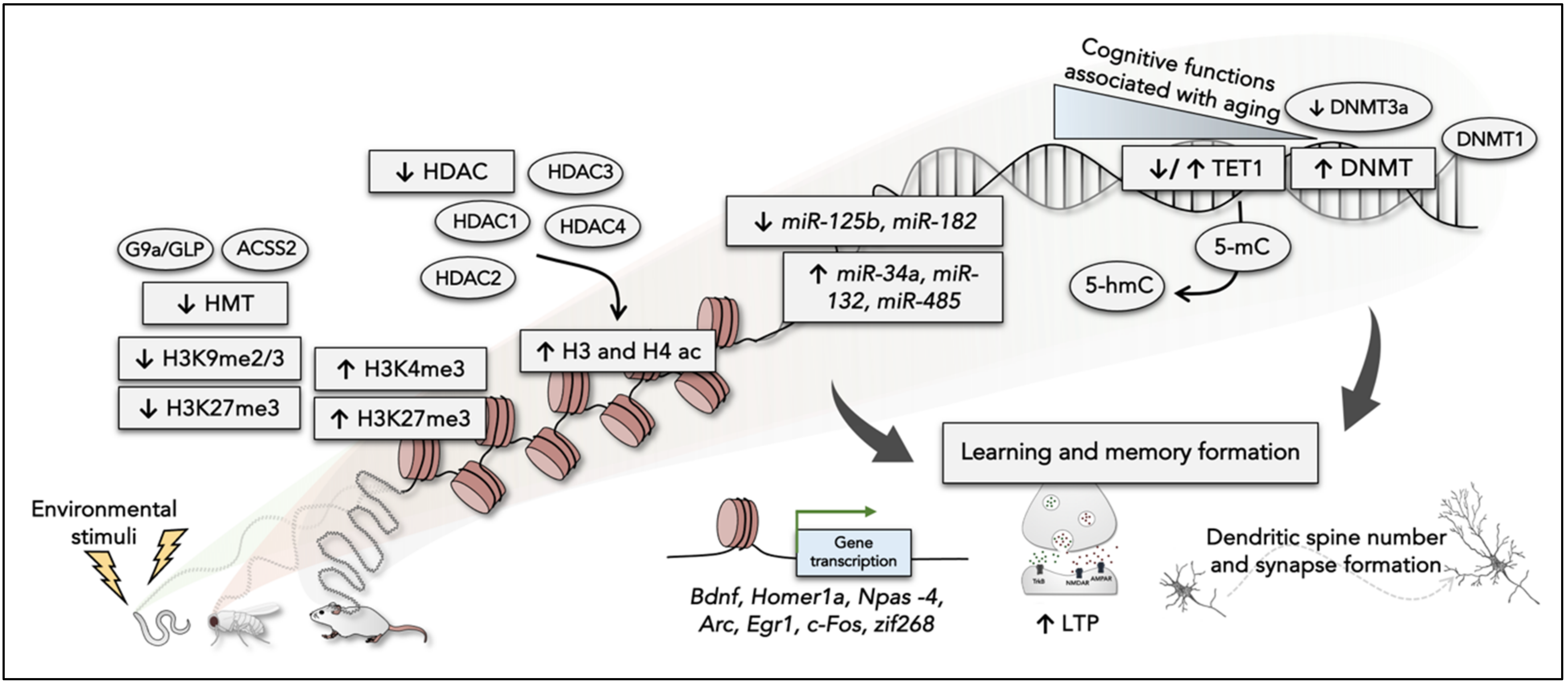
2. The Role of Epigenetic Mechanisms in Learning and Memory Formation
3. Epigenetic Deregulation in Neurodegenerative Diseases: AD as a Model

{kind=link}
{kind=link}
| Epigenetic Mechanism | Epigenetic Alteration | Levels in AD | Model | Outcome | Refs |
|---|---|---|---|---|---|
| DNA methylation | Dnmt1 Dnmt3a | ↓ | DKO mice | Loss of LTP at CA1 synapses in the hippocampus and deficits in hippocampus-based learning and memory | [34] |
| Tet1 | ↓ | C57B6/L mice | Deficit in long-term contextual fear memory | [41] | |
| TREM2 | ↑ | Human | Increased immune genes | [75] | |
| PIN1 | ↓ | Human | Increased AD risk | [76] | |
| TNF-α | ↓ | Human | Encodes multifunctional pro-inflammatory cytokines | [77] | |
| GSK3B | ↑ | Human | Increased Aβ deposition and NFTs | [78] | |
| IL-6 IL-1β | ↑ | Human | Increased inflammatory responses | [79] | |
| APP * | ↓ | Human | Increased Aβ deposition | [78,80,81] | |
| MAPT | ↑ | Human | Increased Tau protein levels | [82] | |
| PSEN1 | ↓ | TgCRND8 mice Human | Increased Aβ deposition | [83,84] | |
| Histone modifications | HDAC1 HDAC2 | ↑ | HDACKO mice Ck-p25 mice Sprague-Dawley rats | Increased Aβ deposition Block expression neuroplasticity genes Reduces the histone acetylation of important genes for learning and memory Decrease of dendritic spine density, synapse number | [47,57,85] |
| HDAC3 | ↑ | HDAC3-Flox mice | Impairment of long-term memory for object recognition | [55] | |
| HDAC4 | ↓ | HDAC4KO mice | Impairment of synaptic plasticity and memory formation | [59] | |
| HDAC6 | ↑ | HDAC6KO mice | Potential modulator of Tau phosphorylation and its aggregations | [86] | |
| SIRT1 | ↓ | N2aSwe/APP cells SIRT-null and SIRT1F/F mice Human | Increased formation of Aβ peptides Downregulation of alpha-secretase ADAM10 Tau protein aggregation | [87,88] | |
| H3K9ac | ↓ | Long-Evans rats | Impairment of learning process | [89] | |
| H3K27ac | ↑↓ | Ck-p25 mice C57BL/6mice Human | Increased immune genes Decreased on synaptic plasticity genes | [90,91] | |
| H4K12ac | ↓ | C57BL/6mice | Age-related memory loss | [92] | |
| H3K4me3 | ↑ | Fischer-344 rats C57BL/6mice | Increased of somatostatin and cortistatin genes Age-related memory decline | [93,94] | |
| H3K9me2 | ↑ | Fischer-344 rats | Decreased Bdnf transcription Age-related memory decline | [93] | |
| H3K36me | ↓ | Age-related memory decline | [95] | ||
| H3K79me | SAMP8 | ||||
| H4K20me | |||||
| miRNA | miR-29 | ↓ | Human | Increased Bace1 expression Increased Aβ deposition | [96] |
| miR-107 | |||||
| miR-132 | ↓ | APP/PS1 mice | Increased Aβ deposition Increased Tau hyperphosphorylation | [97] | |
| miR-138 | ↑ | N2a/APP and HEK293/Tau cells | Increased Tau hyperphosphorylation | [98] | |
| miR-195 | Sprague-Dawley rats | [99] | |||
| miR-206 | ↑ | Tg2576AD mice Human | Downregulation of BDNF gene expression | [100] | |
| miR-132 | ↓ | miR-132/212 KO mice Human | Tau protein overexpression, hyperphosphorylation, and aggregation | [101] | |
| miR-219 | ↓ | D. melanogaster that produces human Tau | Block of repression Tau synthesis | [102] |
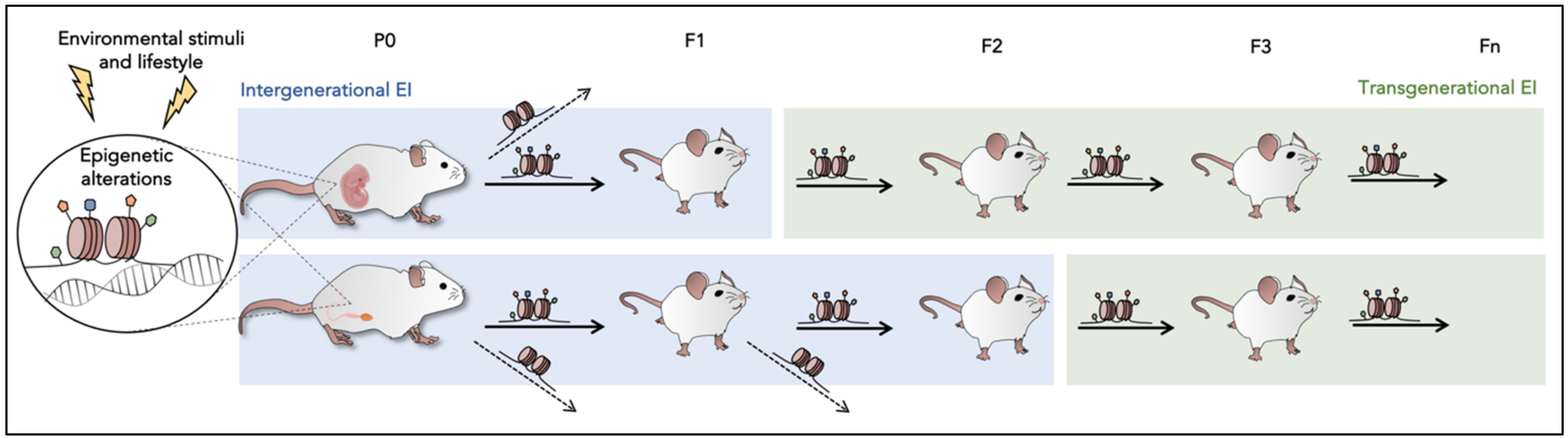
4. Epigenetic Inheritance (EI)
5. Brief Understanding of Mechanism Candidates in EI
6. Evidence in Model Organisms: From One Generation to the Next Generations
7. The Relevant Contribution of TEI in AD Heritability
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2011, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Pishva, E.; Lunnon, K.; van den Hove, D.L. Neuroepigenetics of Aging and Age-Related Neurodegenerative Disorders, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 158, ISBN 9780128125922. [Google Scholar]
- Landgrave-Gómez, J.; Mercado-Gómez, O.; Guevara-Guzmán, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Delgado-Morales, R.; Agís-Balboa, R.C.; Esteller, M.; Berdasco, M. Epigenetic mechanisms during ageing and neurogenesis as novel therapeutic avenues in human brain disorders. Clin. Epigenet. 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M. Mechanisms underlying longevity: A genetic switch model of aging. Exp. Gerontol. 2018, 107, 136–139. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 6, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L. Epigenetic and transgenerational reprogramming of brain development. Nat. Rev. Neurosci. 2015, 16, 332–344. [Google Scholar] [CrossRef]
- Fagiolini, M.; Jensen, C.L.; Champagne, F.A. Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol. 2009, 19, 207–212. [Google Scholar] [CrossRef]
- Day, J.J.; Childs, D.; Guzman-Karlsson, M.C.; Kibe, M.; Moulden, J.; Song, E.; Tahir, A.; Sweatt, J.D. DNA methylation regulates associative reward learning. Nat. Neurosci. 2013, 16, 1445. [Google Scholar] [CrossRef]
- McEwen, B.S.; Eiland, L.; Hunter, R.G.; Miller, M.M. Stress and anxiety: Structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology 2012, 62, 3–12. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Swiergiel, A.H.; Lisowski, P. Epigenetics of stress adaptations in the brain. Brain Res. Bull. 2013, 98, 76–92. [Google Scholar] [CrossRef]
- Cortés-Mendoza, J.; de León-Guerrero, S.D.; Pedraza-Alva, G.; Pérez-Martínez, L. Shaping synaptic plasticity: The role of activity-mediated epigenetic regulation on gene transcription. Int. J. Dev. Neurosci. 2013, 31, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; Anreiter, I.; Aristizabal, M.; Frankland, P.W.; Binder, E.B.; Citri, A. The role of the genome in experience-dependent plasticity: Extending the analogy of the genomic action potential. Proc. Natl. Acad. Sci. USA 2020, 117, 23252–23260. [Google Scholar] [CrossRef] [PubMed]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone methylation regulates memory formation. J. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Griñán-Ferré, C.; Corpas, R.; Puigoriol-Illamola, D.; Palomera-Ávalos, V.; Sanfeliu, C.; Pallàs, M. Understanding Epigenetics in the Neurodegeneration of Alzheimer’s Disease: SAMP8 Mouse Model. J. Alzheimers Dis. 2018, 62, 943–963. [Google Scholar] [CrossRef]
- Day, J.J.; Sweatt, J.D. Cognitive neuroepigenetics: A role for epigenetic mechanisms in learning and memory. Neurobiol. Learn. Mem. 2011, 96, 2–12. [Google Scholar] [CrossRef]
- Khalaf, O.; Gräff, J. Structural, synaptic, and epigenetic dynamics of enduring memories. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef]
- Semon, R.W. Die Mneme als Erhaltendes Prinzip im Wechsel des Organischen Geschehens; Wilhelm Engelmann: Leipzig, Germany, 1911; Volume 7101. [Google Scholar]
- Tonegawa, S.; Pignatelli, M.; Roy, D.S.; Ryan, T.J. Memory engram storage and retrieval. Curr. Opin. Neurobiol. 2015, 35, 101–109. [Google Scholar] [CrossRef]
- Roberson, E.D.; Sweatt, J.D. A biochemical blueprint for long-term memory. Learn. Mem. 1999, 6, 381–388. [Google Scholar] [CrossRef]
- Bae, B.-I.; Jayaraman, D.; Walsh, C.A. Genetic changes shaping the human brain. Dev. Cell 2015, 32, 423–434. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Sarroca, S.; Ivanova, A.; Puigoriol-Illamola, D.; Aguado, F.; Camins, A.; Sanfeliu, C.; Pallàs, M. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging 2016, 8, 664. [Google Scholar] [CrossRef]
- Jablonka, E.; Raz, G. Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef]
- Harper, L. Epigenetic inheritance and the intergenerational transfer of experience. Psychol. Bull. 2005, 131, 340. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Mansuy, I.M. Epigenetic dysregulation in cognitive disorders. Eur. J. Neurosci. 2009, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cassilhas, R.C.; Tufik, S.; de Mello, M.T. Physical exercise, neuroplasticity, spatial learning and memory. Cell. Mol. life Sci. 2016, 73, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Alberini, C.M.; Kandel, E.R. The regulation of transcription in memory consolidation. Cold Spring Harbor Perspect. Biol. 2015, 7, a021741. [Google Scholar] [CrossRef]
- Peixoto, L.; Abel, T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 2013, 38, 62–76. [Google Scholar] [CrossRef]
- Levenson, J.M.; Sweatt, J.D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005, 6, 108–118. [Google Scholar] [CrossRef]
- Koyama, M.; Kurumizaka, H. Structural diversity of the nucleosome. J. Biochem. 2018, 163, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gagnidze, K.; Pfaff, D.W. Epigenetic Mechanisms: DNA Methylation and Histone Protein Modification. In Neuroscience in the 21st Century: From Basic to Clinical; Pfaff, D.W., Ed.; Springer: New York, NY, USA, 2013; pp. 1939–1978. ISBN 978-1-4614-1997-6. [Google Scholar]
- Day, J.J.; Sweatt, J.D. DNA methylation and memory formation. Nat. Neurosci. 2010, 13, 1319–1323. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.-C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef]
- Sultan, F.A.; Wang, J.; Tront, J.; Liebermann, D.A.; Sweatt, J.D. Genetic deletion of Gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity. J. Neurosci. 2012, 32, 17059–17066. [Google Scholar] [CrossRef]
- Zhao, Z.; Fan, L.; Frick, K.M. Epigenetic alterations regulate estradiol-induced enhancement of memory consolidation. Proc. Natl. Acad. Sci. USA 2010, 107, 5605–5610. [Google Scholar] [CrossRef]
- Karaca, K.G.; Kupke, J.; Brito, D.V.C.; Zeuch, B.; Thome, C.; Weichenhan, D.; Lutsik, P.; Plass, C.; Oliveira, A.M.M. Neuronal ensemble-specific DNA methylation strengthens engram stability. Nat. Commun. 2020, 11, 639. [Google Scholar] [CrossRef]
- Oliveira, A.M.M.; Estévez, M.A.; Hawk, J.D.; Grimes, S.; Brindle, P.K.; Abel, T. Subregion-specific p300 conditional knock-out mice exhibit long-term memory impairments. Learn. Mem. 2011, 18, 161–169. [Google Scholar] [CrossRef]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.; King, J.R.; Song, H.; Sweatt, J.D. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef]
- Kumar, D.; Aggarwal, M.; Kaas, G.A.; Lewis, J.; Wang, J.; Ross, D.L.; Zhong, C.; Kennedy, A.; Song, H.; Sweatt, J.D. Tet1 oxidase regulates neuronal gene transcription, active DNA hydroxymethylation, object location memory, and threat recognition memory. Neuroepigenetics 2015, 4, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.-H. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef]
- Monsey, M.S.; Ota, K.T.; Akingbade, I.F.; Hong, E.S.; Schafe, G.E. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PloS ONE 2011, 6, e19958. [Google Scholar] [CrossRef] [PubMed]
- Koshibu, K.; Gräff, J.; Beullens, M.; Heitz, F.D.; Berchtold, D.; Russig, H.; Farinelli, M.; Bollen, M.; Mansuy, I.M. Protein phosphatase 1 regulates the histone code for long-term memory. J. Neurosci. 2009, 29, 13079–13089. [Google Scholar] [CrossRef]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef]
- Kwapis, J.L.; Alaghband, Y.; López, A.J.; White, A.O.; Campbell, R.R.; Dang, R.T.; Rhee, D.; Tran, A.V.; Carl, A.E.; Matheos, D.P. Context and auditory fear are differentially regulated by HDAC3 activity in the lateral and basal subnuclei of the amygdala. Neuropsychopharmacology 2017, 42, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Itzhak, Y.; Anderson, K.L.; Kelley, J.B.; Petkov, M. Histone acetylation rescues contextual fear conditioning in nNOS KO mice and accelerates extinction of cued fear conditioning in wild type mice. Neurobiol. Learn. Mem. 2012, 97, 409–417. [Google Scholar] [CrossRef]
- Bredy, T.W.; Wu, H.; Crego, C.; Zellhoefer, J.; Sun, Y.E.; Barad, M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn. Mem. 2007, 14, 268–276. [Google Scholar] [CrossRef]
- Collins, B.E.; Greer, C.B.; Coleman, B.C.; Sweatt, J.D. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin 2019, 12, 7. [Google Scholar] [CrossRef]
- Bahari-Javan, S.; Maddalena, A.; Kerimoglu, C.; Wittnam, J.; Held, T.; Bähr, M.; Burkhardt, S.; Delalle, I.; Kügler, S.; Fischer, A. HDAC1 regulates fear extinction in mice. J. Neurosci. 2012, 32, 5062–5073. [Google Scholar] [CrossRef] [PubMed]
- Rogge, G.A.; Singh, H.; Dang, R.; Wood, M.A. HDAC3 is a negative regulator of cocaine-context-associated memory formation. J. Neurosci. 2013, 33, 6623–6632. [Google Scholar] [CrossRef]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 2011, 31, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Giustetto, M.; Lomvardas, S.; Kim, J.-H.; Miniaci, M.C.; Schwartz, J.H.; Thanos, D.; Kandel, E.R. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 2002, 111, 483–493. [Google Scholar] [CrossRef]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55. [Google Scholar] [CrossRef]
- Morris, M.J.; Mahgoub, M.; Na, E.S.; Pranav, H.; Monteggia, L.M. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J. Neurosci. 2013, 33, 6401–6411. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Akhtar, M.W.; Adachi, M.; Mahgoub, M.; Bassel-Duby, R.; Kavalali, E.T.; Olson, E.N.; Monteggia, L.M. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci. 2012, 32, 10879–10886. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Agarwal, S.; Franklin, A.V.; DeRamus, T.; Wheelock, M.; Davis, R.L.; McMahon, L.L.; Lubin, F.D. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci. 2012, 32, 5440–5453. [Google Scholar] [CrossRef]
- Alarcón, J.M.; Malleret, G.; Touzani, K.; Vronskaya, S.; Ishii, S.; Kandel, E.R.; Barco, A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: A model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 2004, 42, 947–959. [Google Scholar] [CrossRef]
- Lin, Q.; Wei, W.; Coelho, C.M.; Li, X.; Baker-Andresen, D.; Dudley, K.; Ratnu, V.S.; Boskovic, Z.; Kobor, M.S.; Sun, Y.E. The brain-specific microRNA miR-128b regulates the formation of fear-extinction memory. Nat. Neurosci. 2011, 14, 1115. [Google Scholar] [CrossRef]
- Griggs, E.M.; Young, E.J.; Rumbaugh, G.; Miller, C.A. MicroRNA-182 regulates amygdala-dependent memory formation. J. Neurosci. 2013, 33, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Dias, B.G.; Goodman, J.V.; Ahluwalia, R.; Easton, A.E.; Andero, R.; Ressler, K.J. Amygdala-dependent fear memory consolidation via miR-34a and Notch signaling. Neuron 2014, 83, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Konopka, W.; Kiryk, A.; Novak, M.; Herwerth, M.; Parkitna, J.R.; Wawrzyniak, M.; Kowarsch, A.; Michaluk, P.; Dzwonek, J.; Arnsperger, T. MicroRNA loss enhances learning and memory in mice. J. Neurosci. 2010, 30, 14835–14842. [Google Scholar] [CrossRef]
- Hu, Z.; Li, Z. miRNAs in synapse development and synaptic plasticity. Curr. Opin. Neurobiol. 2017, 45, 24–31. [Google Scholar] [CrossRef]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2018 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2017 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic mechanisms of longevity and aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Smith, A.R.; Smith, R.G.; Condliffe, D.; Hannon, E.; Schalkwyk, L.; Mill, J.; Lunnon, K. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol. Aging 2016, 47, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K. Usefulness of DNA methylation levels in COASY and SPINT1 gene promoter regions as biomarkers in diagnosis of Alzheimer’s disease and amnestic mild cognitive impairment. PLoS ONE 2016, 11, e0168816. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Ramirez, A.; Pieper, H.; Schmitt, I.; Jessen, F.; Wüllner, U. DNA methylation of the TNF-α promoter region in peripheral blood monocytes and the cortex of human Alzheimer’s disease patients. Dement. Geriatr. Cogn. Disord. 2014, 38, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Nagata, K.; Hatsuta, H.; Takuma, H.; Bundo, M.; Iwamoto, K.; Tamaoka, A.; Murayama, S.; Saido, T.; Tsuji, S. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum. Mol. Genet. 2014, 23, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Cavallaro, R.A.; López-González, I.; Maccarrone, M.; Scarpa, S.; Ferrer, I.; Fuso, A. DNA methylation profiles of selected pro-inflammatory cytokines in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2017, 76, 27–31. [Google Scholar] [CrossRef] [PubMed]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region −224 approximately −101 of the amyloid precursor protein gene in autopsy human cortex. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Zhang, C.-C.; Xing, A.; Tan, M.-S.; Tan, L.; Yu, J.-T. The role of MAPT in neurodegenerative diseases: Genetics, mechanisms and therapy. Mol. Neurobiol. 2016, 53, 4893–4904. [Google Scholar] [CrossRef]
- Monti, N.; Cavallaro, R.A.; Stoccoro, A.; Nicolia, V.; Scarpa, S.; Kovacs, G.G.; Fiorenza, M.T.; Lucarelli, M.; Aronica, E.; Ferrer, I. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 2020, 15, 781–799. [Google Scholar] [CrossRef]
- Fuso, A.; Cavallaroa, R.A.; Nicolia, V.; Scarpa, S. PSEN1 promoter demethylation in hyperhomocysteinemic TgCRND8 mice is the culprit, not the consequence. Curr. Alzheimer Res. 2012, 9, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.-S.; Wang, W.-Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.F.; Fass, D.M.; Kao, P.F.; Kahn, M. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.F.; Fletcher, B.R.; Kelley-Bell, B.; Kim, D.H.; Gallagher, M.; Rapp, P.R. Age-related memory impairment is associated with disrupted multivariate epigenetic coordination in the hippocampus. PLoS ONE 2012, 7, e33249. [Google Scholar] [CrossRef]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.-H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef]
- Cheng, H.; Xuan, H.; Green, C.D.; Han, Y.; Sun, N.; Shen, H.; McDermott, J.; Bennett, D.A.; Lan, F.; Han, J.-D.J. Repression of human and mouse brain inflammaging transcriptome by broad gene-body histone hyperacetylation. Proc. Natl. Acad. Sci. USA 2018, 115, 7611–7616. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef]
- Morse, S.J.; Butler, A.A.; Davis, R.L.; Soller, I.J.; Lubin, F.D. Environmental enrichment reverses histone methylation changes in the aged hippocampus and restores age-related memory deficits. Biology 2015, 4, 298–313. [Google Scholar] [CrossRef]
- Rubio, A.; Sánchez-Mut, J.V.; García, E.; Velasquez, Z.D.; Oliver, J.; Esteller, M.; Avila, J. Epigenetic control of somatostatin and cortistatin expression by β amyloid peptide. J. Neurosci. Res. 2012, 90, 13–20. [Google Scholar] [CrossRef]
- Wang, C.M.; Tsai, S.N.; Yew, T.W.; Kwan, Y.W.; Ngai, S.M. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 2010, 11, 87–102. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Salta, E.; Sierksma, A.; Vanden Eynden, E.; De Strooper, B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol. Med. 2016, 8, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, L.; Lu, Y.; Peng, J.; Zhu, Y.; Zhang, Y.; Sun, Z. MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 2015, 589, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ban, T.; Liu, C.; Chen, Q.; Wang, X.U.; Yan, M.; Hu, X.; Su, X.; Bao, Y.; Sun, L. Activation of Cdk5/p25 and tau phosphorylation following chronic brain hypoperfusion in rats involves micro RNA-195 down-regulation. J. Neurochem. 2015, 134, 1139–1151. [Google Scholar] [CrossRef]
- Lee, S.; Chu, K.; Jung, K.; Kim, J.H.; Huh, J.; Yoon, H.; Park, D.; Lim, J.; Kim, J.; Jeon, D. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Alaniz, M.E.; Renwick, N.; Cela, C.; Fulga, T.A.; Van Vactor, D.; Tuschl, T.; Clark, L.N.; Shelanski, M.L.; McCabe, B.D. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J. Clin. Invest. 2015, 125, 681–686. [Google Scholar] [CrossRef]
- Barrachina, M.; Ferrer, I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol. 2009, 68, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Hajkova, P.; Erhardt, S.; Lane, N.; Haaf, T.; El-Maarri, O.; Reik, W.; Walter, J.; Surani, M.A. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 2002, 117, 15–23. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat. Rev. Genet. 2008, 9, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Surani, M.A.; Hayashi, K.; Hajkova, P. Genetic and epigenetic regulators of pluripotency. Cell 2007, 128, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Hajkova, P.; Ancelin, K.; Waldmann, T.; Lacoste, N.; Lange, U.C.; Cesari, F.; Lee, C.; Almouzni, G.; Schneider, R.; Surani, M.A. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 2008, 452, 877–881. [Google Scholar] [CrossRef]
- Morgan, H.D.; Sutherland, H.G.E.; Martin, D.I.K.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar] [CrossRef]
- Lacal, I.; Ventura, R. Epigenetic inheritance: Concepts, mechanisms and perspectives. Front. Mol. Neurosci. 2018, 11, 292. [Google Scholar] [CrossRef]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef]
- Skinner, M.K. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod. Toxicol. 2008, 25, 2–6. [Google Scholar] [CrossRef]
- Perez, M.F.; Lehner, B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat. Cell Biol. 2019, 21, 143–151. [Google Scholar] [CrossRef]
- Benyshek, D.C.; Johnston, C.S.; Martin, J.F.; Ross, W.D. Insulin sensitivity is normalized in the third generation (F3) offspring of developmentally programmed insulin resistant (F2) rats fed an energy-restricted diet. Nutr. Metab. 2008, 5, 26. [Google Scholar] [CrossRef]
- Greer, E.L.; Maures, T.J.; Ucar, D.; Hauswirth, A.G.; Mancini, E.; Lim, J.P.; Benayoun, B.A.; Shi, Y.; Brunet, A. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 2011, 479, 365–371. [Google Scholar] [CrossRef]
- Feng, X.; Guang, S. Small RNAs, RNAi and the inheritance of gene silencing in Caenorhabditis elegans. J. Genet. Genomics 2013, 40, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Tomizawa, S.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811. [Google Scholar] [CrossRef]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Surani, M.A. Germline and pluripotent stem cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a019422. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Dean, W.; Erhardt, S.; Hajkova, P.; Surani, A.; Walter, J.; Reik, W. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 2003, 35, 88–93. [Google Scholar] [CrossRef]
- Radford, E.J.; Ito, M.; Shi, H.; Corish, J.A.; Yamazawa, K.; Isganaitis, E.; Seisenberger, S.; Hore, T.A.; Reik, W.; Erkek, S. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 2014, 345, 1255903. [Google Scholar] [CrossRef]
- Miltenberger, R.J.; Mynatt, R.L.; Wilkinson, J.E.; Woychik, R.P. The role of the agouti gene in the yellow obese syndrome. J. Nutr. 1997, 127, 1902S–1907S. [Google Scholar] [CrossRef] [PubMed]
- Cropley, J.E.; Suter, C.M.; Beckman, K.B.; Martin, D.I.K. CpG methylation of a silent controlling element in the murine A vy allele is incomplete and unresponsive to methyl donor supplementation. PLoS ONE 2010, 5, e9055. [Google Scholar] [CrossRef] [PubMed]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Casas, E.; Vavouri, T. Sperm epigenomics: Challenges and opportunities. Front. Genet. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wang, Q.; Sun, Q.-Y. Histone modifications during mammalian oocyte maturation: Dynamics, regulation and functions. Cell Cycle 2010, 9, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Erkek, S.; Hisano, M.; Liang, C.-Y.; Gill, M.; Murr, R.; Dieker, J.; Schübeler, D.; Van Der Vlag, J.; Stadler, M.B.; Peters, A.H.F.M. Molecular determinants of nucleosome retention at CpG-rich sequences in mouse spermatozoa. Nat. Struct. Mol. Biol. 2013, 20, 868. [Google Scholar] [CrossRef]
- Gaydos, L.J.; Wang, W.; Strome, S. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science 2014, 345, 1515–1518. [Google Scholar] [CrossRef]
- Siklenka, K.; Erkek, S.; Godmann, M.; Lambrot, R.; McGraw, S.; Lafleur, C.; Cohen, T.; Xia, J.; Suderman, M.; Hallett, M. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 2015, 350, aab2006. [Google Scholar] [CrossRef]
- Yan, W. Potential roles of noncoding RNAs in environmental epigenetic transgenerational inheritance. Mol. Cell. Endocrinol. 2014, 398, 24–30. [Google Scholar] [CrossRef]
- Ashe, A.; Sapetschnig, A.; Weick, E.-M.; Mitchell, J.; Bagijn, M.P.; Cording, A.C.; Doebley, A.-L.; Goldstein, L.D.; Lehrbach, N.J.; Le Pen, J. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell 2012, 150, 88–99. [Google Scholar] [CrossRef]
- Rechavi, O.; Houri-Ze’evi, L.; Anava, S.; Goh, W.S.S.; Kerk, S.Y.; Hannon, G.J.; Hobert, O. Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 2014, 158, 277–287. [Google Scholar] [CrossRef]
- Grandjean, V.; Fourré, S.; De Abreu, D.A.F.; Derieppe, M.-A.; Remy, J.-J.; Rassoulzadegan, M. RNA-mediated paternal heredity of diet-induced obesity and metabolic disorders. Sci. Rep. 2015, 5, 18193. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.; Peng, H.; Zhang, X.; Zhang, Y. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gapp, K.; Jawaid, A.; Sarkies, P.; Bohacek, J.; Pelczar, P.; Prados, J.; Farinelli, L.; Miska, E.; Mansuy, I.M. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 2014, 17, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cozen, A.E.; Liu, Y.; Chen, Q.; Lowe, T.M. Small RNA modifications: Integral to function and disease. Trends Mol. Med. 2016, 22, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Galan, C.; Krykbaeva, M.; Rando, O.J. Early life lessons: The lasting effects of germline epigenetic information on organismal development. Mol. Metab. 2020, 38, 100924. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.; Schneper, L.M.; Notterman, D.A. DNA methylation, early life environment, and health outcomes. Pediatr. Res. 2016, 79, 212–219. [Google Scholar] [CrossRef]
- Liu, D.; Diorio, J.; Day, J.C.; Francis, D.D.; Meaney, M.J. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat. Neurosci. 2000, 3, 799–806. [Google Scholar] [CrossRef]
- Houri-Zeevi, L.; Kohanim, Y.K.; Antonova, O.; Rechavi, O. Three rules explain transgenerational small RNA inheritance in C. elegans. Cell 2020, 182, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, O.; Lev, I. Principles of transgenerational small RNA inheritance in Caenorhabditis elegans. Curr. Biol. 2017, 27, R720–R730. [Google Scholar] [CrossRef]
- Moore, R.S.; Kaletsky, R.; Murphy, C.T. Piwi/PRG-1 Argonaute and TGF-β Mediate Transgenerational Learned Pathogenic Avoidance. Cell 2019, 177, 1827–1841.e12. [Google Scholar] [CrossRef]
- Woodhouse, R.M.; Buchmann, G.; Hoe, M.; Harney, D.J.; Low, J.K.K.; Larance, M.; Boag, P.R.; Ashe, A. Chromatin Modifiers SET-25 and SET-32 Are Required for Establishment but Not Long-Term Maintenance of Transgenerational Epigenetic Inheritance. Cell Rep. 2018, 25, 2259–2272.e5. [Google Scholar] [CrossRef] [PubMed]
- Klosin, A.; Casas, E.; Hidalgo-Carcedo, C.; Vavouri, T.; Lehner, B. Transgenerational transmission of environmental information in C. elegans. Science 2017, 356, 320–323. [Google Scholar] [CrossRef]
- Rechtsteiner, A.; Ercan, S.; Takasaki, T.; Phippen, T.M.; Egelhofer, T.A.; Wang, W.; Kimura, H.; Lieb, J.D.; Strome, S. The Histone H3K36 Methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- Katz, D.J.; Edwards, T.M.; Reinke, V.; Kelly, W.G. A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell 2009, 137, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.H.; Li, D.; Shimizu, H.; Nakamura, R.; Ishii, S. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 2011, 145, 1049–1061. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Maternal E-Cigarette Exposure Results in Cognitive and Epigenetic Alterations in Offspring in a Mouse Model. Chem. Res. Toxicol. 2018, 31, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Donath, A.; Yarmolenko, M.; Uslar, E.; Conzen, C.; Kanakis, D.; Bosma, C.; Worm, K.; Paulus, W.; Keyvani, K. Exercise during pregnancy mitigates Alzheimer-like pathology in mouse offspring. FASEB J. 2012, 26, 117–128. [Google Scholar] [CrossRef]
- Bordoni, L.; Nasuti, C.; Di Stefano, A.; Marinelli, L.; Gabbianelli, R. Epigenetic Memory of Early-Life Parental Perturbation: Dopamine Decrease and DNA Methylation Changes in Offspring. Oxid. Med. Cell. Longev. 2019, 2019, 1472623. [Google Scholar] [CrossRef]
- Modir, F.; Salmani, M.E.; Goudarzi, I.; Lashkarboluki, T.; Abrari, K. Prenatal stress decreases spatial learning and memory retrieval of the adult male offspring of rats. Physiol. Behav. 2014, 129, 104–109. [Google Scholar] [CrossRef]
- Lucia, D.; Burgess, D.; Cullen, C.L.; Dorey, E.S.; Rawashdeh, O.; Moritz, K.M. Periconceptional maternal alcohol consumption leads to behavioural changes in adult and aged offspring and alters the expression of hippocampal genes associated with learning and memory and regulators of the epigenome. Behav. Brain Res. 2019, 362, 249–257. [Google Scholar] [CrossRef]
- Wimmer, M.E.; Briand, L.A.; Fant, B.; Guercio, L.A.; Arreola, A.C.; Schmidt, H.D.; Sidoli, S.; Han, Y.; Garcia, B.A.; Pierce, R.C. Paternal cocaine taking elicits epigenetic remodeling and memory deficits in male progeny. Mol. Psychiatry 2017, 22, 1641–1650. [Google Scholar] [CrossRef]
- Roth, T.L.; Lubin, F.D.; Funk, A.J.; Sweatt, J.D. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 2009, 65, 760–769. [Google Scholar] [CrossRef]
- Bohacek, J.; Mansuy, I.M. Epigenetic inheritance of disease and disease risk. Neuropsychopharmacology 2013, 38, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Xavier, M.J.; Roman, S.D.; Aitken, R.J.; Nixon, B. Transgenerational inheritance: How impacts to the epigenetic and genetic information of parents affect offspring health. Hum. Reprod. Update 2019, 25, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Escott-Price, V.; Shoai, M.; Pither, R.; Williams, J.; Hardy, J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol. Aging 2017, 49, 214.e7–214.e11. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Crow, T.J. The missing genes: What happened to the heritability of psychiatric disorders? Mol. Psychiatry 2011, 16, 362–364. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef]
- Mill, J. Toward an integrated genetic and epigenetic approach to Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1188–1191. [Google Scholar] [CrossRef]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef]
- Yu, J.-T.; Xu, W.; Tan, C.-C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef]
- Youngson, N.A.; Whitelaw, E. Transgenerational epigenetic effects. Annu. Rev. Genom. Hum. Genet. 2008, 9, 233–257. [Google Scholar] [CrossRef] [PubMed]
- Griñán-Ferré, C.; Izquierdo, V.; Otero, E.; Puigoriol-Illamola, D.; Corpas, R.; Sanfeliu, C.; Ortuño-Sahagún, D.; Pallàs, M. Environmental enrichment improves cognitive deficits, AD hallmarks and epigenetic alterations presented in 5xFAD mouse model. Front. Cell. Neurosci. 2018, 12, 224. [Google Scholar] [CrossRef] [PubMed]
- Griñan-Ferré, C.; Puigoriol-Illamola, D.; Palomera-Ávalos, V.; Pérez-Cáceres, D.; Companys-Alemany, J.; Camins, A.; Ortuño-Sahagún, D.; Rodrigo, M.T.; Pallàs, M. Environmental enrichment modified epigenetic mechanisms in SAMP8 mouse hippocampus by reducing oxidative stress and inflammaging and achieving neuroprotection. Front. Aging Neurosci. 2016, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Yeshurun, S.; Hannan, A.J. Transgenerational epigenetic influences of paternal environmental exposures on brain function and predisposition to psychiatric disorders. Mol. Psychiatry 2019, 24, 536–548. [Google Scholar] [CrossRef]
- Rachdaoui, N.; Sarkar, D.K. Transgenerational epigenetics and brain disorders. Int. Rev. Neurobiol. 2014, 115, 51–73. [Google Scholar]
- Branchi, I. The mouse communal nest: Investigating the epigenetic influences of the early social environment on brain and behavior development. Neurosci. Biobehav. Rev. 2009, 33, 551–559. [Google Scholar] [CrossRef]
- Fenoglio, K.A.; Brunson, K.L.; Baram, T.Z. Hippocampal neuroplasticity induced by early-life stress: Functional and molecular aspects. Front. Neuroendocrinol. 2006, 27, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Molteni, R.; Racagni, G.; Riva, M.A. Stress during development: Impact on neuroplasticity and relevance to psychopathology. Prog. Neurobiol. 2007, 81, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Ickes, B.R.; Pham, T.M.; Sanders, L.A.; Albeck, D.S.; Mohammed, A.H.; Granholm, A.-C. Long-term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp. Neurol. 2000, 164, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, F.; Zhuang, Y.; Feng, J.; Ying, Z.; Fan, G. Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur. J. Neurosci. 2011, 33, 383–390. [Google Scholar] [CrossRef]
- Izquierdo, V.; Palomera-Ávalos, V.; Pallàs, M.; Griñán-Ferré, C. Resveratrol Supplementation Attenuates Cognitive and Molecular Alterations under Maternal High-Fat Diet Intake: Epigenetic Inheritance over Generations. Int. J. Mol. Sci. 2021, 22, 1453. [Google Scholar] [CrossRef]
- Izquierdo, V.; Palomera-Ávalos, V.; López-Ruiz, S.; Canudas, A.M.; Pallàs, M.; Griñán-Ferré, C. Maternal resveratrol supplementation prevents cognitive decline in senescent mice offspring. Int. J. Mol. Sci. 2019, 20, 1134. [Google Scholar] [CrossRef]


| Model | M or P Inh | Experimental Design | Mechanism Lo-of-Function | Epigenetic Alteration | Up to | Outcomes | Refs |
|---|---|---|---|---|---|---|---|
| C. elegans | M,P | Learned behavior avoidance of pathogenic bacteria | Piwi/PRG-1 | F4 | TEI of Pseudomonas aeruginosa avoidance. | [143] | |
| M,P | Gene silencing | set-25 and set-32 mutation | H3K9me3 | F3 | RNAi-induced TEI involves initiation of silencing by canonical RNAi pathway genes, establishing heritable silencing by set-25 and set-32, and ongoing maintenance of heritable silencing requiring small RNA-associated genes such as hrde-1 and nrde-2. | [144] | |
| M,P | Temperature-sensitive transcriptional repression during 5 generations | set-25 mutation | H3K9me2/3 | F14 | Reactivation of SET-25-silenced transposons. Inheritance occurs through both oocytes and sperm. | [145] | |
| M,P | Gene silencing | Hrde-1/Wago-9, and, set-25 and set-32 | piRNAs | F24 | Germline nuclear small RNA/chromatin pathway can maintain stable inheritance for many generations when triggered by a piRNA-dependent foreign RNA response. | [132] | |
| M,P | Epigenetic memory | mes-4 mutation | H3K36me | F1 | MES-4 transmits the memory of gene expression in the parental germline to offspring, and that this memory role is critical for the PGCs to execute a proper germline program. | [146] | |
| M,P | Epigenetic memory | Spr-5 (KDM1) mutation | H3K4me2 | F30 | The progressive derepression of genes that regulate spermatogenesis, defects in oogenesis and spermatogenesis and sterility | [147] | |
| D. melanogaster | M,P | Heterochromatin organization | High-temperature induced p-Atf-2 | H3K9me2 | F5 | Reduction of H3K9me2, disruption of heterochromatin formation and gene silencing | [148] |
| Model | M or P Inh | Experimental Design | Mechanism Loss-of-Function | Epigenetic Alteration | Up to | Outcomes | Refs |
|---|---|---|---|---|---|---|---|
| Balb/C mice | M | Intergenerational transmission of aversive exposure attenuates Cognitive and Molecular | E-Cigarette exposure | DNA methylation | F1 | Significant changes in global DNA methylation associated with significant changes in chromatin modification enzymes in the brains of the offspring. Maternal exposure to e-cigarette aerosols resulted in both cognitive and epigenetic changes in offspring were found. | [149] |
| CRND8 mice | M | Exercise during pregnancy | Early-life exposure | DNA methylation | F1 | Exercise during pregnancy provides long-lasting protection from neurodegeneration and improves brain plasticity in the otherwise unstimulated progeny. | [150] |
| Wistar rats | M,P | Epigenetic memory | Early-life exposure to permethrin | 5-mC 5-hmC | F1 | Since the F1 generation did not receive any permethrin, the impairments observed in DNA methylation and hydroxymethylation, together with a reduction in dopamine levels in the F1 generation, have to be associated with parental early-life exposure to permethrin. | [151] |
| M | Epigenetic reprogramming | Early life or prenatal stress induces | DNA methylation | F4 | HSS decreased learning and memory of adult offspring in BPS and PS1, prominently. | [152] | |
| Sprague-Dawley rats | M | Intergenerational transmission of alcohol consumption | Early exposure to alcohol | DNMT1 DNMT3a HDAC2 | F1 | Alcohol around the time of conception leads to sex and age specific behavioral adaptations later in life, along with gene expression changes to the methyltransferases, histone modifiers and other genes important for learning and memory. | [153] |
| P | Epigenetic reprogramming | Exposure to cocaine | H3K4me1 H3ac | F1 | Epigenetic changes in the hippocampus of male progeny associated with open chromatin states were found. | [154] | |
| Long-Evans rats | M | Epigenetic reprogramming | Early life or prenatal stress induces | DNA methylation | F1 | Early maltreatment produced persisting changes in methylation of BDNF DNA that caused altered BDNF gene expression in the adult prefrontal cortex. Altered BDNF DNA methylation in offspring of females that had previously experienced the maltreatment regimen. | [140,155] |
| Model | M or P Inh | Experimental Design | Mechanism Loss-of-Function | Epigenetic Alteration | Up To | Outcomes | Refs |
|---|---|---|---|---|---|---|---|
| SAMP8 mice | M | Intergenerational transmission of diet attenuates Cognitive and Molecular | HFD | 5-mC Dnmt1 Dnmt3a m6A | F2 | A significant increase in DNA methylation levels. Significant increase of m6A levels in HFD+RSV F1 and changes in gene expression of its enzymes Mettl3 and Fto. | [179] |
| M | Intergenerational transmission of diet attenuates Cognitive and Molecular | Supplementary diet | 5-mC/5-hmC Dnmt3A/B Tet2 | F2 | Maternal resveratrol supplementation could prevent cognitive impairment in the SAMP8 mice offspring through epigenetic changes and cell signaling pathways. | [180] | |
| CRND8 mice | M | Exercise during pregnancy | Early-life exposure | DNA methylation | F1 | Exercise during pregnancy provides long-lasting protection from neurodegeneration and improves brain plasticity in the otherwise unstimulated progeny. | [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellver-Sanchis, A.; Pallàs, M.; Griñán-Ferré, C. The Contribution of Epigenetic Inheritance Processes on Age-Related Cognitive Decline and Alzheimer’s Disease. Epigenomes 2021, 5, 15. https://doi.org/10.3390/epigenomes5020015
Bellver-Sanchis A, Pallàs M, Griñán-Ferré C. The Contribution of Epigenetic Inheritance Processes on Age-Related Cognitive Decline and Alzheimer’s Disease. Epigenomes. 2021; 5(2):15. https://doi.org/10.3390/epigenomes5020015
Chicago/Turabian StyleBellver-Sanchis, Aina, Mercè Pallàs, and Christian Griñán-Ferré. 2021. "The Contribution of Epigenetic Inheritance Processes on Age-Related Cognitive Decline and Alzheimer’s Disease" Epigenomes 5, no. 2: 15. https://doi.org/10.3390/epigenomes5020015
APA StyleBellver-Sanchis, A., Pallàs, M., & Griñán-Ferré, C. (2021). The Contribution of Epigenetic Inheritance Processes on Age-Related Cognitive Decline and Alzheimer’s Disease. Epigenomes, 5(2), 15. https://doi.org/10.3390/epigenomes5020015