An Epigenetics-Based Hypothesis of Autoantigen Development in Systemic Lupus Erythematosus


Abstract
1. Introduction—The Complexity of Autoimmune Diseases
2. Components of the “X Chromosome-Nucleolus Nexus” Hypothesis
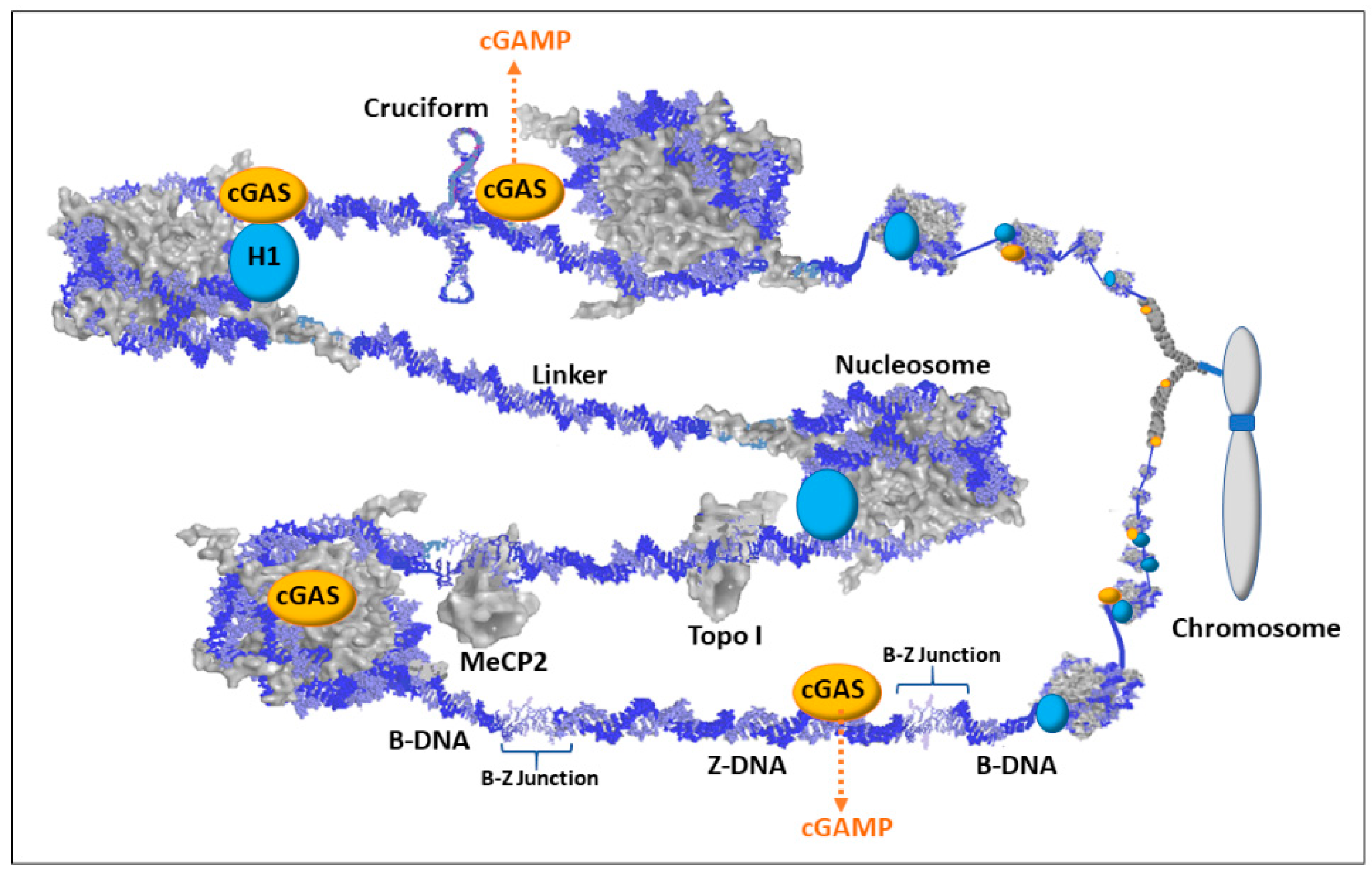
2.1. The Hypothesis in Brief
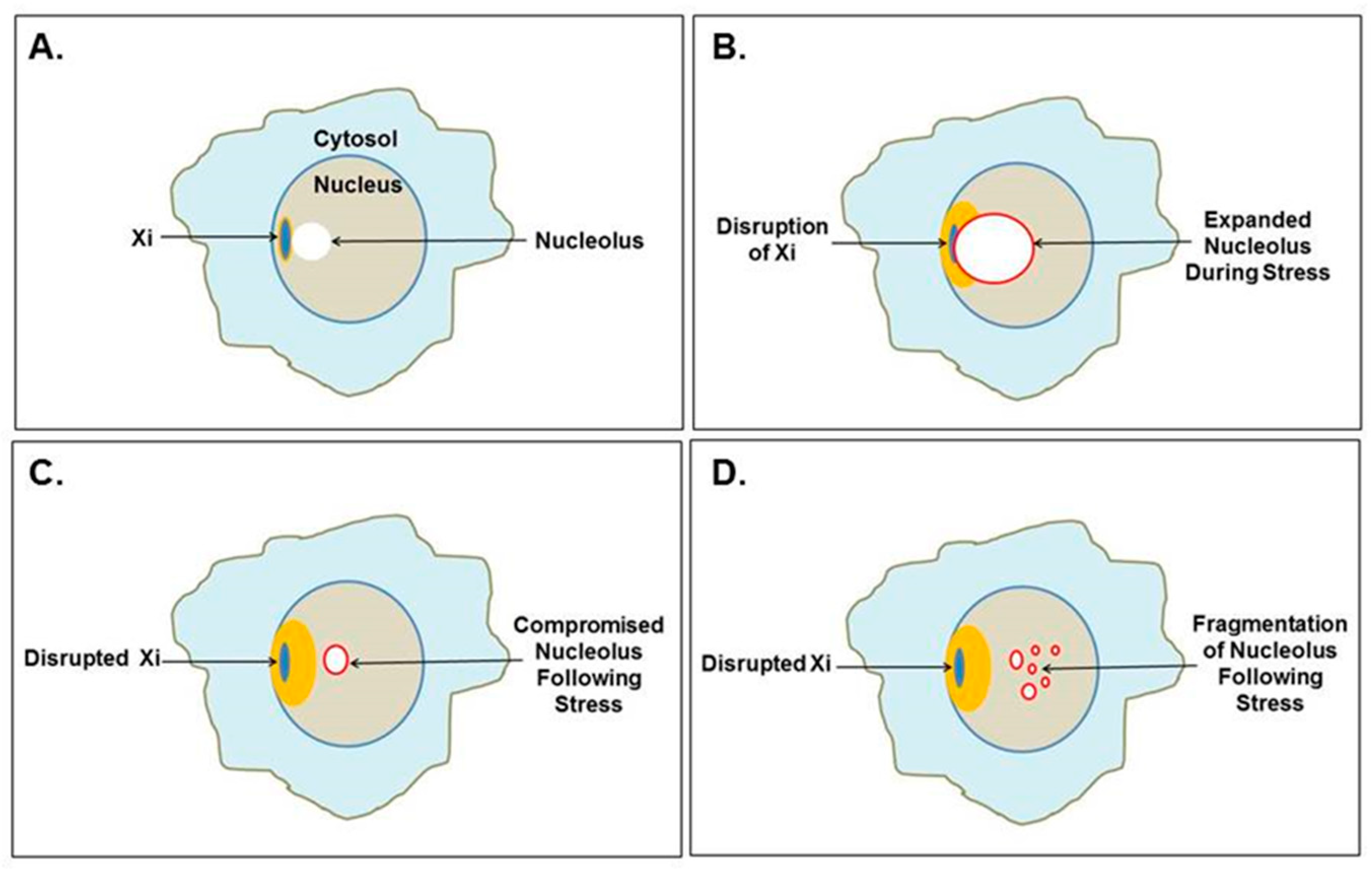
2.2. The Nucleolus: Functions and Stress
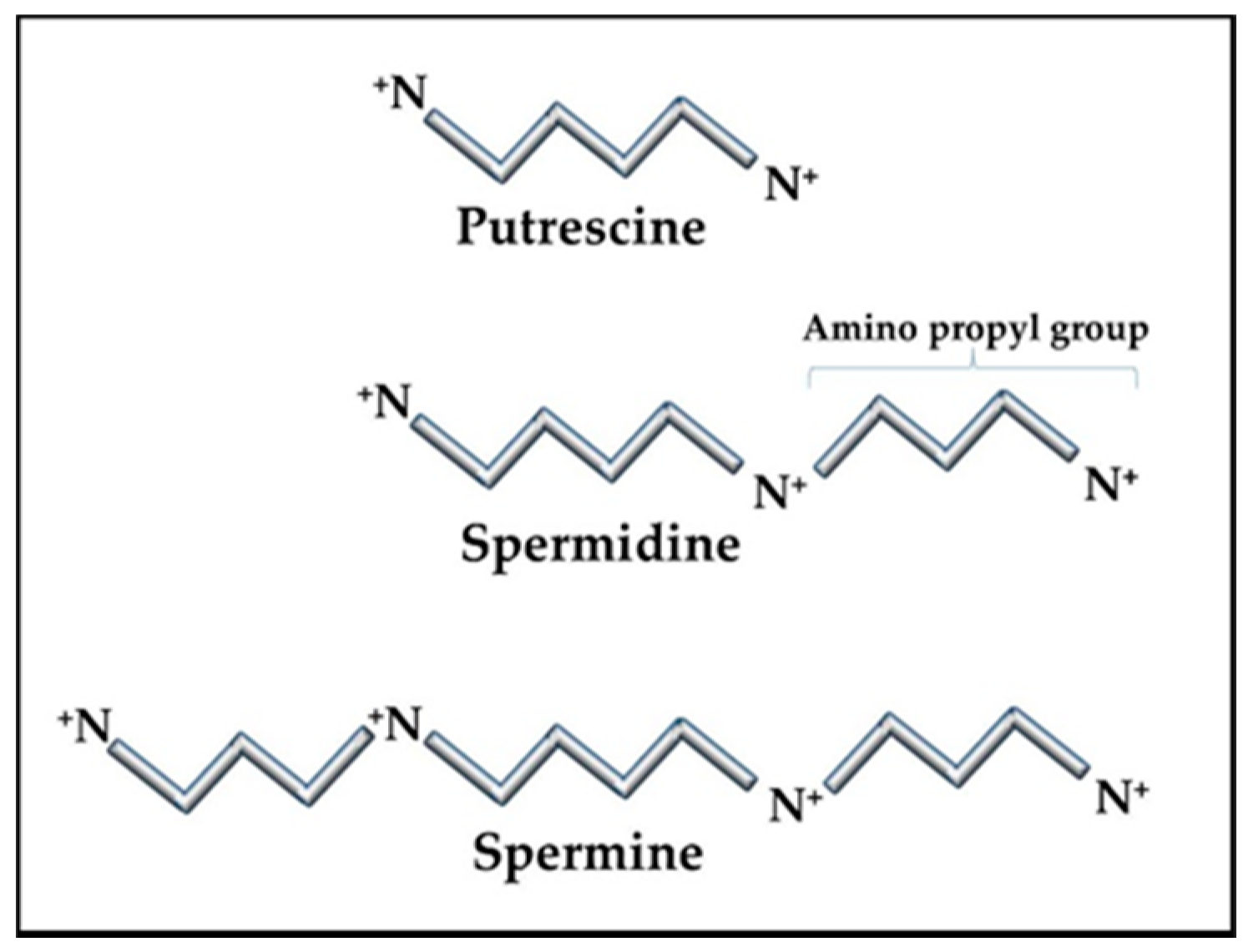
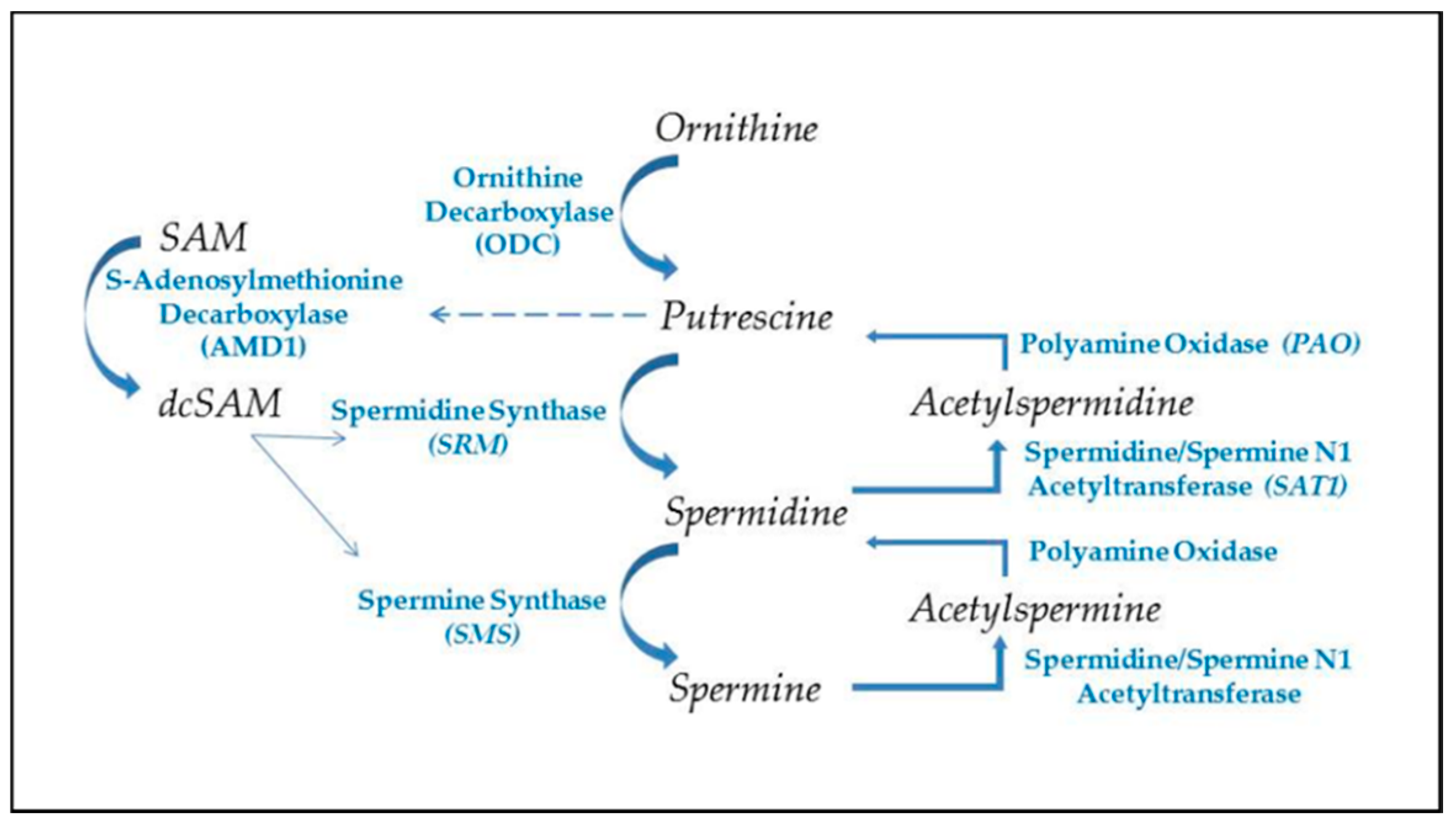
2.3. Polyamines
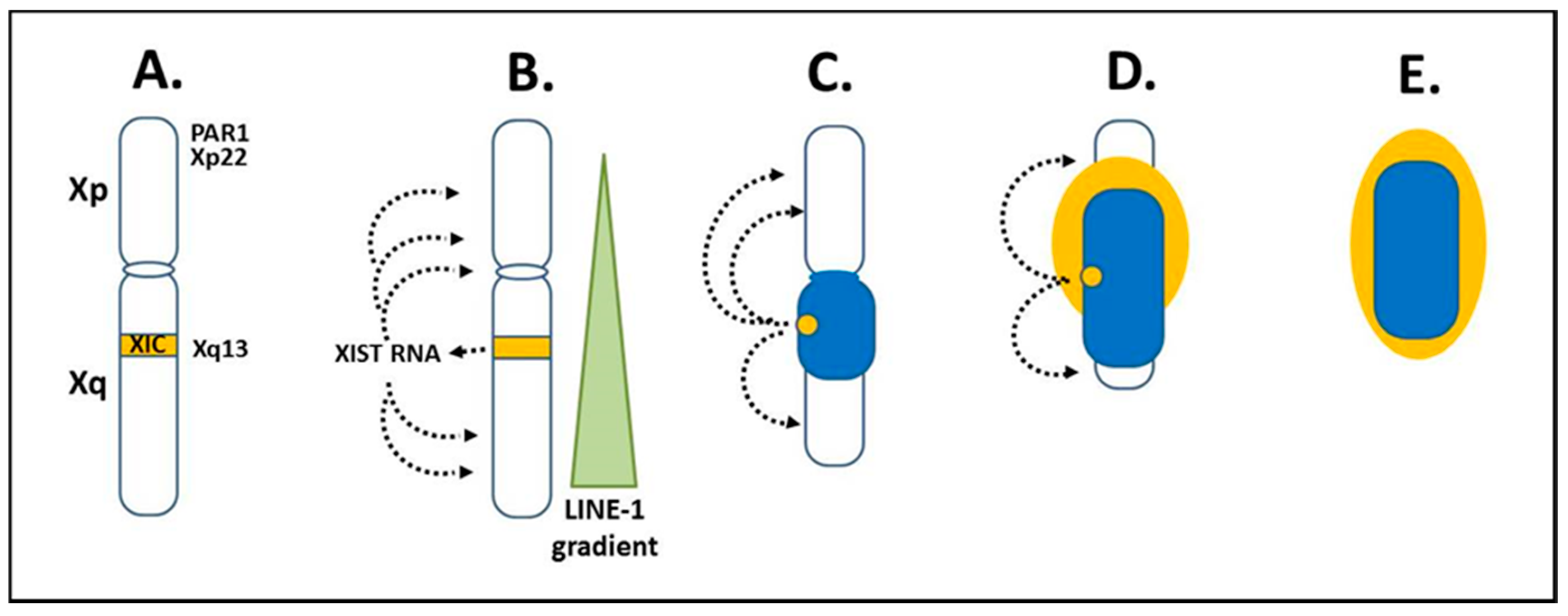
2.4. The Inactive X Chromosome
3. The “X Chromosome-Nucleolus Nexus” Hypothesis in Action
3.1. Disruption of the Inactive X Chromosome
3.2. Disruption of the Nucleolus
3.3. New Developments in the “X Chromosome-Nucleolus Nexus” Hypothesis
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- American Autoimmune Related Diseases Association. Available online: https://www.aarda.org/diseaselist/ (accessed on 9 May 2019).
- D’Andrea, M.R. Add Alzheimer’s disease to the list of autoimmune diseases. Med. Hypotheses 2005, 64, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Madrid, F.F.; Maroun, M.C.; Olivero, O.A.; Long, M.; Stark, A.; Grossman, L.I.; Binder, W.; Dong, J.; Burke, M.; Nathanson, S.D.; et al. Autoantibodies in breast cancer sera are not epiphenomena and may participate in carcinogenesis. BMC Cancer 2015, 15, 1407–1421. [Google Scholar] [CrossRef]
- Bei, R.; Masuelli, L.; Palumbo, C.; Modesti, M.; Modesti, A. A common repertoire of autoantibodies is shared by cancer and autoimmune disease patients: Inflammation in their induction and impact on tumor growth. Cancer Lett. 2009, 281, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Hajiabbasi, A.; Masooleh, I.S.; Alizadeh, Y.; Banikarimi, A.S.; Ghavidel, P. Secondary Sjogren’s Syndrome in 83 Patients with Rheumatoid Arthritis. Acta Med. Iran. 2016, 54, 448–453. [Google Scholar] [PubMed]
- Manoussakis, M.N.; Georgopoulou, C.; Zintzaras, E.; Spyropoulou, M.; Stavropoulou, A.; Skopouli, F.N.; Moutsopoulos, H.M. Sjögren’s syndrome associated with systemic lupus erythematosus: Clinical and laboratory profiles and comparison with primary Sjögren’s syndrome. Arthritis Rheum. 2004, 50, 882–891. [Google Scholar] [CrossRef]
- De Seze, J.; Devos, D.; Castelnovo, G.; Labauge, P.; Dubucquoi, S.; Stojkovic, T.; Ferriby, D.; Vermersch, P. The prevalence of Sjögren syndrome in patients with primary progressive multiple sclerosis. Neurology 2001, 57, 1359–1363. [Google Scholar] [CrossRef]
- Rojas-Villarraga, A.; Amaya-Amaya, J.; Rodriguez-Rodriguez, A.; Mantilla, R.D.; Anaya, J.M. Introducing polyautoimmunity: Secondary autoimmune diseases no longer exist. Autoimmune Dis. 2011, 2012, 1–9. [Google Scholar] [CrossRef]
- Yaniv, G.; Twig, G.; Shor, D.B.; Furer, A.; Sherer, Y.; Mozes, O.; Komisar, O.; Slonimsky, E.; Klang, E.; Lotan, E.; et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: A diversity of 180 different antibodies found in SLE patients. Autoimmun. Rev. 2015, 14, 75–79. [Google Scholar] [CrossRef]
- Sibley, J.T.; Lee, J.S.; Decoteau, W.E. Left-handed “Z” DNA antibodies in rheumatoid arthritis and systemic lupus erythematosus. J. Rheumatol. 1984, 11, 633–637. [Google Scholar]
- Harley, J.B.; Scofield, R.H.; Reichlin, M. Anti-Ro in Sjögren’s syndrome and systemic lupus erythematosus. Rheum. Dis. Clin. N. Am. 1992, 18, 337–358. [Google Scholar]
- Shen, L.; Suresh, L.; Lindemann, M.; Xuan, J.; Kowal, P.; Malyavantham, K.; Ambrus, J.L., Jr. Novel autoantibodies in Sjogren’s syndrome. Clin. Immunol. 2012, 145, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gershwin, M.E.; Chang, C. Diagnostic criteria for systemic lupus erythematosus: A critical review. J. Autoimmun. 2014, 48–49, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, S.K.; Johnson, S.R.; Boumpas, D.; Daikh, D.; Dörner, T.; Jayne, D.; Kamen, D.; Lerstom, K.; Mosca, M.; Ramsey-Goldman, R.R.; et al. Developing and refining new candidate criteria for systemic lupus erythematosus classification: An international collaboration. Arthritis Care Res. 2018, 70, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H.; Renaudineau, Y. Epigenetics and autoimmune diseases: The X chromosome-nucleolus nexus. Front. Genet. 2015, 6, 1–22. [Google Scholar] [CrossRef]
- Brooks, W.H. Viral impact in autoimmune diseases: Expanding the “X chromosome-nucleolus nexus” hypothesis. Front. Immunol. 2017, 8, 1657. [Google Scholar] [CrossRef]
- Brooks, W.H.; Renaudineau, Y. The ‘nucleolus’ hypothesis of autoimmune diseases and its implications. Eur. Med. J. 2017, 2, 82–89. [Google Scholar]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus and systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 370516, 1–10. [Google Scholar] [CrossRef]
- Yung, R.; Chang, S.; Hemati, N.; Johnson, K.; Richardson, B. Mechanisms of drug-induced lupus. IV. Comparison of procainamide and hydralazine with analogs in vitro and in vivo. Arthritis Rheum. 1997, 40, 1436–1443. [Google Scholar] [CrossRef]
- Zhang, L.F.; Huynh, K.D.; Lee, J.T. Perinucleolar targeting of the inactive X during S phase: Evidence for a role in the maintenance of silencing. Cell 2007, 129, 693–706. [Google Scholar] [CrossRef]
- Barr, M.L.; Bertram, E.G. A morphological distinction between neurons of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. In Problems of Birth Defects; Persaud, T.V.N., Ed.; Springer: Dordrecht, The Netherlands, 1949. [Google Scholar]
- Ohno, S.; Hauschka, T.S. Allocycly of the X-chromosome in tumors and normal tissues. Cancer Res. 1960, 20, 541–545. [Google Scholar]
- Lyon, M.F. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet. 1962, 14, 135–148. [Google Scholar] [PubMed]
- Baranello, L.; Levens, D.; Kouzine, F. DNA supercoiling (omics). In Nuclear Architecture and Dynamics; Translational Epigenetics; Lavelle, C., Victor, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 2, pp. 81–99. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Pankert, T.; Seiler, J.; Németh, A.; Voit, R.; Grummt, I.; Rippe, K. Alu element-containing RNAs maintain nucleolar structure and function. EMBO J. 2015, 34, 2758–2774. [Google Scholar] [CrossRef] [PubMed]
- Reichlin, M. Systemic lupus erythematosus. In Systemic Autoimmunity; Bigazzi, P.E., Reichlin, M., Eds.; Marcel Dekker: New York, NY, USA, 1991; Volume 54, pp. 163–200. [Google Scholar]
- Nagai, T.; Arinuma, Y.; Yanagida, T.; Yamamoto, K.; Hirohata, S. Anti-ribosomal P protein antibody in human systemic lupus erythematosus up-regulates the expression of proinflammatory cytokines by human peripheral blood monocytes. Arthritis Rheum. 2005, 52, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Lassoued, K.; Guilly, M.N.; Danon, F.; Andre, C.; Dhumeaux, D.; Clauve, J.P.; Brouet, J.C.; Seligmann, M.; Courvalin, J.C. Antinuclear autoantibodies specific for lamins: Characterization and clinical significance. Ann. Intern. Med. 1988, 108, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.S.; Jurada, D.; Bursac, S.; Orsolic, I.; Bartek, J.; Volarevic, S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.W.; Trinkle-Mulcahy, L. New insights into nucleolar structure and function. F1000Prime Rep. 2015, 7, 48–57. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L. Nucleolus: The consummate nuclear body. In Nuclear Architecture and Dynamics; Translational Epigenetics; Academic Press: Cambridge, MA, USA, 2018; Volume 2, pp. 257–282. [Google Scholar] [CrossRef]
- Latonen, L. Phase-to-phase with nucleoli—Stress responses, protein aggregation and novel roles of RNA. Front. Cell. Neurosci. 2019, 13, 151. [Google Scholar] [CrossRef]
- Boisvert, F.M.; Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Hernandez-Verdun, D.; Roussel, P.; Thiery, M.; Sirri, V.; Lafontaine, D.L.J. The nucleolus: Structure/function relationship in RNA metabolism. Wiley Interdiscip. Rev. RNA 2010, 1, 415–431. [Google Scholar] [CrossRef]
- Dubois, M.L.; Boisvert, F.M. The nucleolus: Structure and function. In The Functional Nucleus; Bazett-Jones, D.P., Gellaire, G., Eds.; Springer Intnational Publication: Cham, Switzerland, 2016; pp. 29–49. [Google Scholar] [CrossRef]
- Padeken, J.; Heun, P. Nucleolus and nuclear periphery: Velcro for heterochromatin. Curr. Opin. Cell Biol. 2014, 28, 54–60. [Google Scholar] [CrossRef]
- Grumml, I. The nucleolus—The guardian of cellular homeostasis and genome integrity. Chromosoma 2013, 122, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Paule, M.R.; White, R.J. Survey and summary: Transcription by RNA polymerases I and III. Nucleic Acids Res. 2000, 28, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Keenan, R.J.; Freymann, D.M.; Stroud, R.M.; Walter, P. The signal recognition particle. Ann. Rev. Biochem. 2001, 70, 755–775. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Catalano, A. Proteins of the nucleolus: An introduction. In Proteins of the Nucleolus; O’Day, D., Catalano, A., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 3–15. [Google Scholar]
- Ahmad, Y.; Boisvert, F.M.; Gregor, P.; Cobley, A.; Lamond, A.I. NOPdb: Nucleolar proteome database—2008 update. Nucleic Acids Res. 2009, 37, D181–D184. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Nakamuta, H.; Oda-Ueda, N.; Larsson, L.I.; Fujiwara, K. Immunocytochemical demonstration of polyamines in nucleoli and nuclei. Histochem. Cell. Biol. 2008, 129, 659–665. [Google Scholar] [CrossRef]
- Gfeller, E.; Stern, D.N.; Russell, D.H.; Levy, C.C.; Taylor, R.L. Ultrastructural changes in vitro of rat liver nucleoli in response to polyamines. Z. Zellforsch. Mikrosk. Anat. 1972, 129, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, R. The proto-oncogene c-myc is a direct target of gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef]
- Dang, C.V. c-Myc targets genes involved in cell growth, apoptosis and metabolism. Mol. Cell Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Weiger, T.M.; Hermann, A. Cell proliferation, potassium channels, polyamines and their interactions: A mini review. Amino Acids 2014, 46, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M. Arginine metabolism revisited. J. Nutr. 2016, 146, 2579S–2586S. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Ealick, S.E. Structural biology of S-adenosylmethionine decarboxylase. Amino Acids 2010, 38, 451–460. [Google Scholar] [CrossRef]
- Hougaard, D.M.; Del Castillo, A.M.; Larsson, L.I. Endogenous polyamines associate with DNA during its condensation in mammalian tissue. A fluorescence cytochemical and immunocytochemical study of polyamines in fetal rat liver. Eur. J. Cell Biol. 1988, 45, 311–314. [Google Scholar]
- Xaplanteri, M.A.; Petropoulos, A.D.; Dinos, G.P.; Kalpaxis, D.L. Localization of spermine binding sites in 23S rRNA by photoaffinity labeling: Parsing the spermine contribution to ribosomal 50S subunit functions. Nucleic Acids Res. 2005, 33, 2792–2805. [Google Scholar] [CrossRef] [PubMed]
- Jovine, L.; Djordjevic, S.; Rhodes, D. The crystal structure of yeast phenylalanine tRNA at 2.0 Å resolution: Cleavage by Mg2+ in 15-year old crystals. J. Mol. Biol. 2000, 301, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Ahmed, K.; Even-Faitelson, L.; Lleres, D.; Bazett-Jones, D.P.; Lamond, A.I. Modulation of higher order chromatin conformation in mammalian cell nuclei can be mediated by polyamines and divalent cations. PLoS ONE 2013, 8, e67689. [Google Scholar] [CrossRef]
- Rich, A.; Zhang, S. Timeline: Z-DNA: The long road to biological function. Nat. Rev. Genet. 2003, 4, 566–572. [Google Scholar] [CrossRef]
- Lilley, D.M.J.; Sullivan, K.M.; Murchie, A.I.H.; Furlong, J.C. Cruciform extrusion in supercoiled DNA—Mechanisms and contextual influence. In Unusual DNA Structures; Wells, R.D., Harvey, S.C., Eds.; Springer: New York, NY, USA, 1988; pp. 55–72. [Google Scholar] [CrossRef]
- Whelly, S.M. Role of polyamine in the regulation of RNA synthesis in uterine nucleoli. J. Steroid Biochem. Mol. Biol. 1991, 39, 161–167. [Google Scholar] [CrossRef]
- Löwkvist, B.; Emanuelsson, H.; Heby, O. Effects of polyamine limitation on nucleolar development and morphology in early chick embryos. Cell Diff. 1983, 12, 19–26. [Google Scholar] [CrossRef]
- Pegg, A.E. Regulation of ornithine decarboxylase. J. Biol. Chem. 2006, 281, 14529–14532. [Google Scholar] [CrossRef]
- Pegg, A.E. S-adenosylmethionine decarboxylase. Essays Biochem. 2009, 46, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Bachman, A.S.; Geerts, D. Polyamine synthesis as a target of MYC oncogenes. J. Biol. Chem. 2018, 293, 18757–18769. [Google Scholar] [CrossRef] [PubMed]
- Gerner, E.W.; Kurtts, T.A.; Fuller, D.J.M.; Casero, R.A. Stress induction of the spermidine/spermine N1-acetyltransferase by a post-transcriptional mechanism in mammalian cells. Biochem. J. 1993, 294, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H. X chromosome inactivation and autoimmunity. Clin. Rev. Allerg. Immunol. 2010, 39, 20–29. [Google Scholar] [CrossRef]
- Stewart, T.M.; Dunston, T.T.; Woster, P.M.; Casero, R.A. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef]
- Stevens, J.F.; Maier, C.S. Acrolein: Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 2008, 52, 7–25. [Google Scholar] [CrossRef]
- Mandal, S.; Mandal, A.; Park, M.H. Depletion of the polyamines spermidine and spermine by overexpression of spermidine/spermine N1-acetyltransferase (SAT1) leads to mitochondria-mediated apoptosis in mammalian cells. Biochem. J. 2015, 468, 435–447. [Google Scholar] [CrossRef]
- Furumitsu, Y.; Yukioka, K.; Kojima, A.; Yukioka, M.; Shichikawa, K.; Ochi, T.; Matsui-Yuasa, I.; Otani, S.; Nishizawa, Y.; Morii, H. Levels of urinary polyamines in patients with rheumatoid arthritis. J. Rheumatol. 1993, 20, 1661–1665. [Google Scholar]
- Karouzakis, E.; Gay, R.E.; Gay, S.; Neidhart, M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012, 64, 1809–1817. [Google Scholar] [CrossRef]
- Neidhart, M.; Karouzakis, E.; Jüngel, A.; Gay, R.E.; Gay, S. Inhibition of spermidine/spermine N1-acetyltransferase activity: A new therapeutic concept in rheumatoid arthritis. Arthritis Rheum. 2014, 66, 1723–1733. [Google Scholar] [CrossRef]
- Thomas, T.J.; Messner, R.P. Difluoromethylornithine therapy of female NZB/W mice. J. Rheum. 1991, 18, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Lee, H.S.; Shin, T.H.; Jung, J.Y.; Baek, W.Y.; Park, H.J.; Lee, G.; Paik, M.J.; Suh, C.H. Polyamine patterns in plasma of patients with systemic lupus erythematosus and fever. Lupus 2018, 27, 930–938. [Google Scholar] [CrossRef]
- Thomas, T.J.; Meryhew, N.L.; Messner, R.P. Enhanced binding of lupus sera to the polyamine-induced left-handed z-dna form of polynucleotides. Arthritis Rheum. 1990, 33, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Yoshida, M.; Igarashi, A.; Ito, K.; Wada, Y.; Murakami, S.; Kobayashi, D.; Nakano, M.; Sohda, M.; Nakajima, T.; et al. Intense correlation between protein-conjugated acrolein and primary Sjögren’s syndrome. Clin. Chim. Acta 2010, 411, 359–363. [Google Scholar] [CrossRef]
- Hirose, T.; Saiki, R.; Uemura, T.; Suzuki, T.; Dohmae, N.; Ito, S.; Takahashi, H.; Ishii, I.; Toida, T.; Kashiwagi, K.; et al. Increase in acrolein-conjugated immunoglobulins in saliva from patients with primary Sjögren’s syndrome. Clin. Chim. Acta 2015, 450, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, A.H.; Harley, J.B.; Scofield, R.H. Autoimmunity and Klinefelter’s syndrome: When men have two X chromosomes. J. Autoimmun. 2009, 33, 31–34. [Google Scholar] [CrossRef]
- Galupa, R.; Heard, E. X-chromosome inactivation: New insights into cis and trans regulation. Curr. Opin. Genet. Dev. 2015, 31, 57–66. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Heard, E. Fifty years of X inactivation research. Development 2011, 138, 5049–5055. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Heard, E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Ann. Rev. Cell Dev. Biol. 2014, 30, 561–580. [Google Scholar] [CrossRef]
- Valencia, K.; Wutz, A. Recent insights into the regulation of X-chromosome inactivation. Adv. Genom. Genet. 2015, 5, 227–238. [Google Scholar] [CrossRef]
- Balaton, B.P.; Dixon-McDougall, T.; Peeters, S.B.; Brown, C.J. The eXceptional nature of the X chromosome. Hum. Mol. Genet. 2018, 27, R242–R249. [Google Scholar] [CrossRef] [PubMed]
- Finestra, T.R.; Gribnau, J. X chromosome inactivation: Silencing, topology and reactivation. Curr. Opin. Cell Biol. 2017, 46, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.G.; Heard, E. Role and control of X chromosome dosage in mammalian development. Curr. Opin. Genet. Dev. 2013, 23, 109–115. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pessia, E.; Makino, T.; Bailly-Bechet, M.; McLysaght, A.; Marais, G.A.B. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc. Natl. Acad. Sci. USA 2012, 109, 5346–5351. [Google Scholar] [CrossRef]
- Bacher, C.P.; Guggiari, M.; Brors, B.; Augui, S.; Clerc, P.; Avner, P.; Eils, R.; Heard, E. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat. Cell Biol. 2006, 8, 293–299. [Google Scholar] [CrossRef]
- Lyon, M.F. X-chromosome inactivation: A repeat hypothesis. Cytogenet. Cell Genet. 1998, 80, 133–137. [Google Scholar] [CrossRef]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howell, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef]
- Helbig, R.; Fackelmayer, F.O. Scaffold attachment factor A (SAF-A) is concentrated in inactive X chromosome territories through its RGG domain. Chromosoma 2003, 112, 173–182. [Google Scholar] [CrossRef]
- Wang, C.Y.; Jegu, T.; Chu, H.P.; Oh, H.J.; Lee, J.T. SMCHD1 merges chromosome compartments and assists formation of super-structures on the inactive X. Cell 2018, 174, 406–421. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.T.; Wang, Y.H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genom. 2015, 16, 60–70. [Google Scholar] [CrossRef]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef]
- Kryczek, I.; Liu, R.; Wang, G.; Wu, K.; Shu, X.; Szeliga, W.; Vatan, L.; Finlayson, E.; Huang, E.; Simeone, D.; et al. FOXP3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Res. 2009, 69, 3995–4000. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, A.; Gorelik, G.; Patel, D.; Liyanarachchi, P.; McCune, W.J.; Somers, E.; Gonzalez-Rivera, T.; Cohort, T.M.; Strickland, F.; Richardson, B. Overexpression of X-linked genes in T cells from women with lupus. J. Autoimmun. 2013, 41, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Konsta, O.D.; Le Dantec, C.; Brooks, W.H.; Renaudineau, Y. Genetics and Epigenetics of Autoimmune Diseases; John Wiley & Sons: Chichester, UK, 2015. [Google Scholar] [CrossRef]
- Perl, A. LINEing up to boost interferon production: Activation of endogenous retroviral DNA in autoimmunity. Arthritis Rheum. 2016, 68, 2568–2570. [Google Scholar] [CrossRef]
- Koelsch, K.A.; Webb, R.; Jeffries, M.; Dozmorov, M.G.; Frank, M.B.; Guthridge, J.M.; James, J.A.; Wren, J.D.; Sawalha, A.H. Functional characterization of the MECP2/IRAK1 lupus risk haplotype in human T cells and a human MECP2 transgenic mouse. J. Autoimmun. 2013, 41, 168–174. [Google Scholar] [CrossRef][Green Version]
- Jacob, C.O.; Zhu, J.; Armstrong, D.L.; Yan, M.; Han, J.; Zhou, X.J.; Thomas, J.A.; Reiff, A.; Myones, B.L.; Ojwang, J.O.; et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2009, 106, 6256–6261. [Google Scholar] [CrossRef]
- Lyu, G.; Tan, T.; Guan, Y.; Sun, L.; Liang, Q.; Tao, W. Changes in the position and volume of inactive X chromosomes during the G0/G1 transition. Chromosome Res. 2018, 26, 179–189. [Google Scholar] [CrossRef]
- Kim, T.H.; Jeon, Y.J.; Yi, J.M.; Kim, D.S.; Huh, J.W.; Hur, C.G.; Kim, H.S. The distribution and expression of HERV families in the human genome. Mol. Cells 2004, 18, 87–93. [Google Scholar]
- Le Dantec, C.; Vallet, S.; Brooks, W.H.; Renaudineau, Y. Human endogenous retrovirus group E and its involvement in diseases. Viruses 2015, 7, 1238–1257. [Google Scholar] [CrossRef]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Steinman, C.R. Plasma DNA in systemic lupus erythematosus. Arthritis Rheum. 1989, 32, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Rubin, C.M.; Schmid, C.W. Genome-wide chromatin remodeling modulates the Alu heat shock response. Gene 2001, 276, 127–133. [Google Scholar] [CrossRef]
- Lanzavecchia, A. How can cryptic epitopes trigger autoimmunity? J. Exp. Med. 1995, 181, 1945–1948. [Google Scholar] [CrossRef]
- Brooks, W.H. Autoimmune diseases and polyamines. Clin. Rev. Allergy Immunol. 2012, 42, 58–70. [Google Scholar] [CrossRef]
- Matsuyama, A.; Croce, C.M.; Huebner, K. Common fragile genes. Eur. J. Histochem. 2004, 48, 29–36. [Google Scholar] [CrossRef]
- Glover, T.W. Common fragile sites. Cancer Lett. 2006, 232, 4–12. [Google Scholar] [CrossRef]
- Dall, K.L.; Scarpini, C.G.; Roberts, I.; Winder, D.M.; Stanley, M.A.; Muralidhar, B.; Herdman, M.T.; Pett, M.R.; Coleman, N. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res. 2008, 68, 8249–8259. [Google Scholar] [CrossRef]
- Sun, Z.; Chadwick, B.P. Loss of SETDB1 decompacts the inactive X chromosome in part through reactivation of an enhancer in the IL1RAPL1 gene. Epigenetics Chromatin 2018, 11, 45–64. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Gulen, M.F. The role of cGAS in innate immunity and beyond. J. Mol. Med. 2016, 94, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kirsch, M.A.; Gong, M.; Chowdhury, D.; Senenko, L.; Engel, K.; Lee, Y.A.; de Silva, U.; Bailey, S.L.; Witte, T.; Vyse, T.J.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007, 39, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 2019, 8, e47491. [Google Scholar] [CrossRef] [PubMed]
- Gekara, N.O.; Jiang, H. The innate immune DNA sensor cGAS: A membrane, cytosolic, or nuclear protein? Sci. Signal. 2019, 12, eaax3521. [Google Scholar] [CrossRef] [PubMed]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.F.; Lombardi, E.P.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. 2019, 26, 2377–2393. [Google Scholar] [CrossRef]
- Wang, A.H.J.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; van Boom, J.H.; van der Marel, G.; Rich, A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef]
- Takeshita, F.; Leifer, C.A.; Gursel, I.; Ishii, K.J.; Takeshita, S.; Gursel, M.; Klinman, D.M. Cutting edge: Role of Toll-like Receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 2001, 167, 3555–3558. [Google Scholar] [CrossRef]
- Zhang, Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Brooks, W.H. Increased polyamines alter chromatin and stabilize autoantigens in autoimmune diseases. Front. Immunol. 2013, 4, 91. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autoantigen | Function/Complex | Occurrence (% in SLE) | Reference |
|---|---|---|---|
| Nucleolin | Nucleolar structural integrity | >50 | [26] |
| U1RNP | Spliceosome component | 40 | [26] |
| U1RNA | Spliceosome component | <5 | [26] |
| Sm epitopes | Spliceosome proteins | 25 | [26] |
| SSA/Ro | RNA pol III chaperone | 40–50 | [26] |
| SSB/La | RNA pol III chaperone and termination | 15 | [26] |
| Ribosomal P proteins | Phospho proteins, bind 28S RNA | 12–16 | [27] |
| Ku | dsDNA break repair | 20–40 | [26] |
| Cardiolipin | Similar epitopes to nucleophosmin | 20–40 | [26] |
| Centromere components | CENP-B and others | ~6 | [26] |
| Lamins | Complexed with nucleolin | unknown | [28] |
| Gene/Site | Name | Location | Potential Issue | Reference |
|---|---|---|---|---|
| Alu elements | Short Interspersed Elements | Enriched in PAR1 | Disruption | [15] |
| FRAXB | Fragile Site B | Xp22 | Latent viruses, DNA damage | [89] |
| (hot) LINE-1 | Long Interspersed Elements | Xp22 | Reverse transcription | [15] |
| SMS | Spermine Synthase | Xp22 | Polyamine dysregulation | [15] |
| SAT1 | Spermidine/Spermine N1 Acetyltransferase | Xp22 | Polyamine dysregulation | [15] |
| TLR7 | Toll-like Receptor 7 | Xp22 | Overexpression | [90] |
| FOXP3 | Forkhead Box P3 | Xp11 | T-cell dysregulation | [91] |
| CXCR3 | C-X-C motif chemokine receptor 3 | Xq13 | Overexpression | [92] |
| FRAXC | Fragile Site C | Xq22 | Latent viruses, DNA damage | [89] |
| CD40L | Cluster of differentiation 40 ligand | Xq24 | Overexpression | [90] |
| HERV-w | Human endogenous retrovirus w | Xq22 | Dysregulation | [93,94] |
| FRAXD | Fragile Site D | Xq27 | Latent viruses, DNA damage | [89] |
| MeCP2 | Methyl-CpG-binding protein 2 | Xq28 | DNA methylation dysregulation | [95] |
| IRAK1 | Interleukin-1 receptor associated kinase-1 | Xq28 | Checkpoint dysregulation | [96] |
| FRAXA | Fragile Site A | Xq28 | Latent viruses, DNA damage | [89] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, W. An Epigenetics-Based Hypothesis of Autoantigen Development in Systemic Lupus Erythematosus. Epigenomes 2020, 4, 6. https://doi.org/10.3390/epigenomes4020006
Brooks W. An Epigenetics-Based Hypothesis of Autoantigen Development in Systemic Lupus Erythematosus. Epigenomes. 2020; 4(2):6. https://doi.org/10.3390/epigenomes4020006
Chicago/Turabian StyleBrooks, Wesley. 2020. "An Epigenetics-Based Hypothesis of Autoantigen Development in Systemic Lupus Erythematosus" Epigenomes 4, no. 2: 6. https://doi.org/10.3390/epigenomes4020006
APA StyleBrooks, W. (2020). An Epigenetics-Based Hypothesis of Autoantigen Development in Systemic Lupus Erythematosus. Epigenomes, 4(2), 6. https://doi.org/10.3390/epigenomes4020006