The Dynamic Partnership of Polycomb and Trithorax in Brain Development and Diseases


Abstract
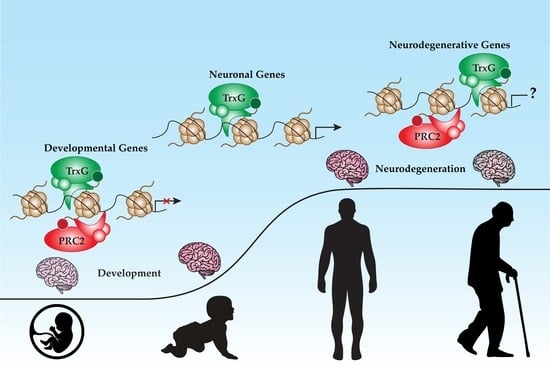
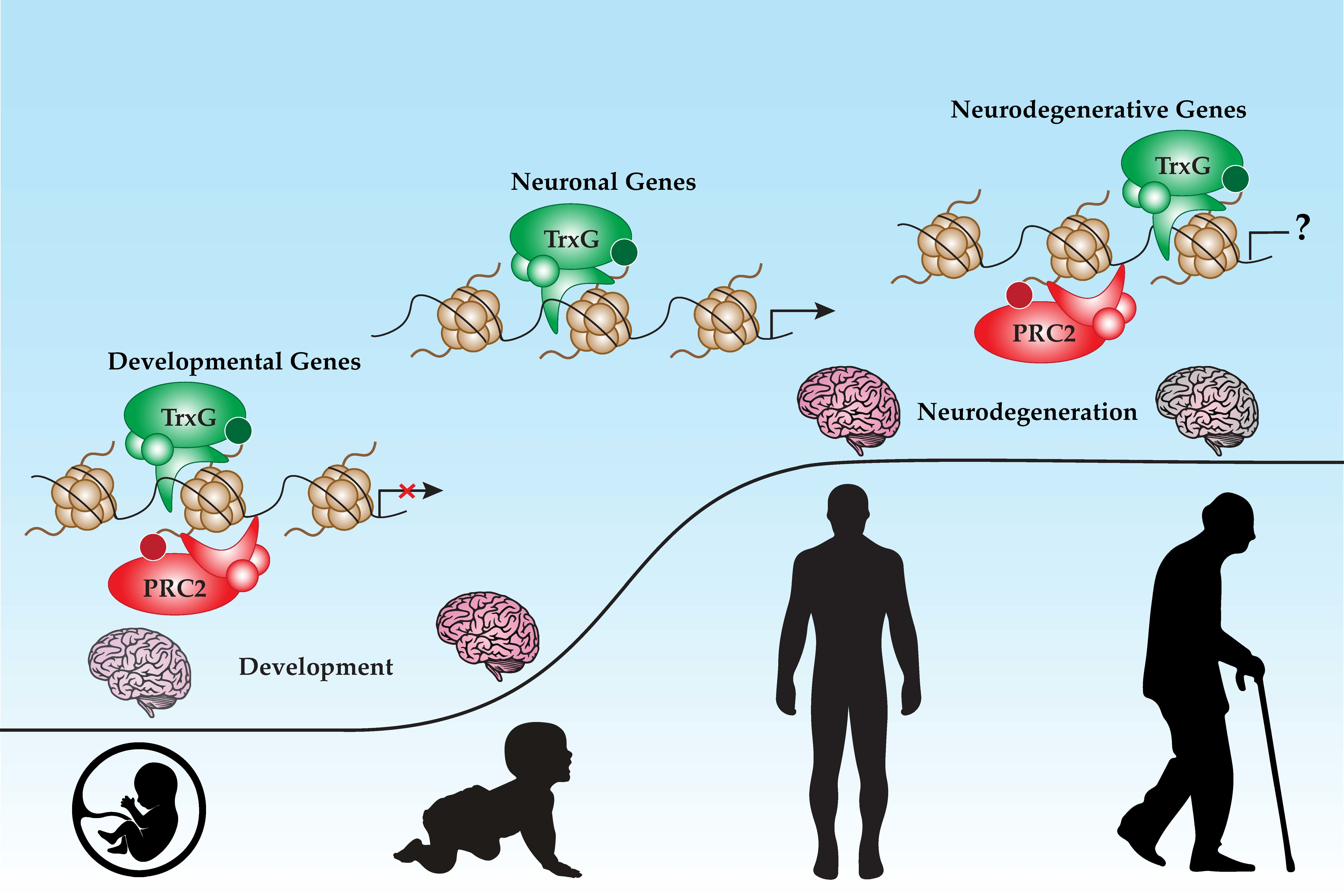{kind=link}
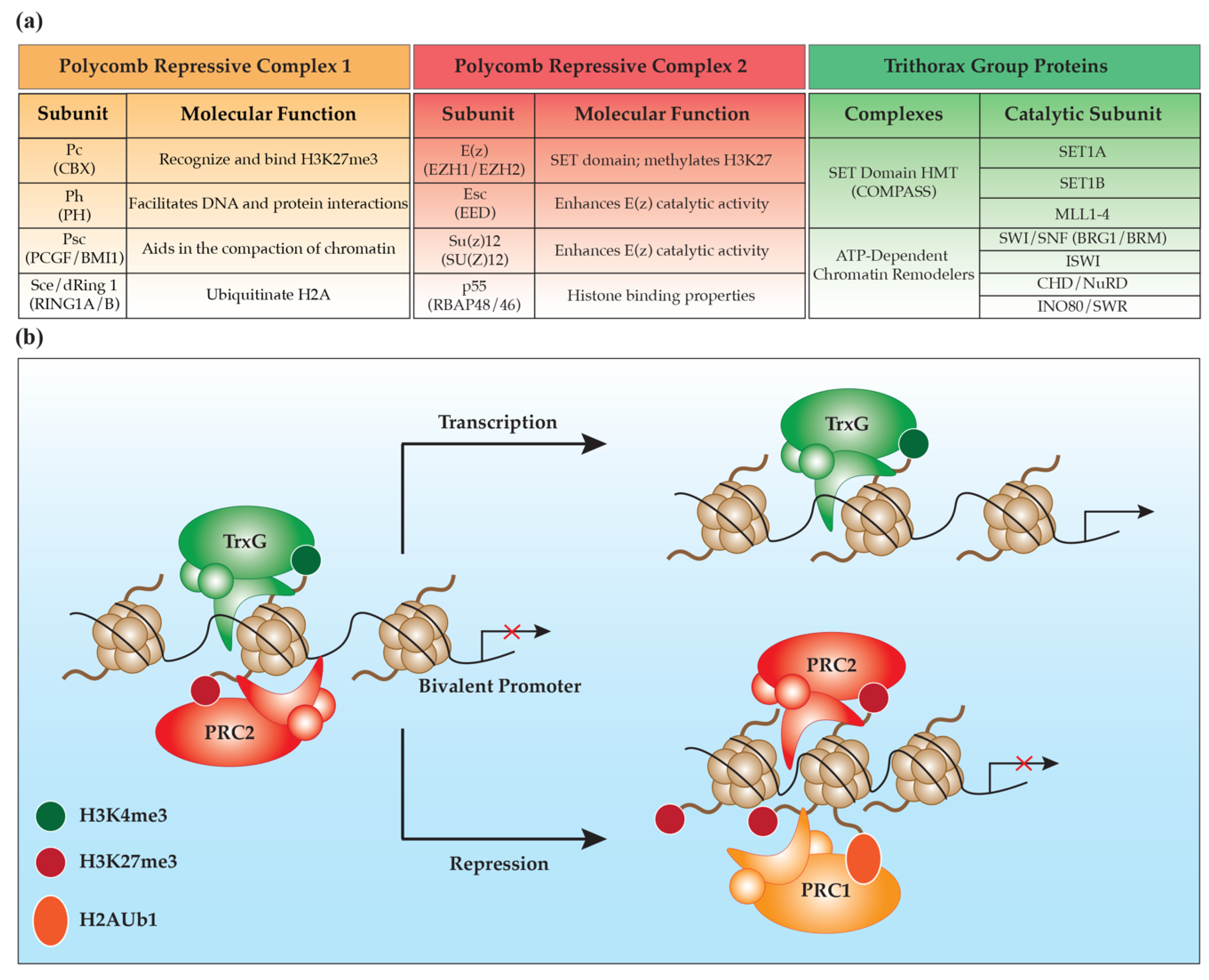{kind=link}
1. Introduction
2. Polycomb Group Proteins
2.1. Polycomb Repressive Complex 2
2.2. Polycomb Repressive Complex 1
3. Trithorax Group Proteins
3.1. Trithorax SET Domain Histone Methyltransferases
3.2. ATP-Dependent Chromatin Remodelers
3.3. TrxG Response Elements (TREs)
4. PcG and TrxG Proteins in the CNS
4.1. PRC1 in Neurogenesis and CNS Development
4.2. PRC2 in Neurogenesis and CNS Development
4.3. TrxG in Neurogenesis and CNS Development
4.4. PcG and TrxG in Astrogliogenesis and Oligodendrogenesis
4.5. PcG and TrxG in Neuronal Migration
4.6. PcG and TrxG in Neuroprotection and Aging
5. PcG and TrxG in Neurodegenerative Diseases
5.1. Huntington’s Disease
5.2. Alzheimer’s Disease
5.3. Parkinson’s Disease
6. Future Outlook and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Chinwalla, V.; Jane, E.P.; Harte, P.J. The Drosophila trithorax protein binds to specific chromosomal sites and is co-localized with Polycomb at many sites. EMBO J. 1995, 14, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. Author Correction: The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2018, 19, 771. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Kageyama, R. Regulation of temporal properties of neural stem cells and transition timing of neurogenesis and gliogenesis during mammalian neocortical development. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Siebold, A.P.; Banerjee, R.; Tie, F.; Kiss, D.L.; Moskowitz, J.; Harte, P.J. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Flamier, A.; El Hajjar, J.; Adjaye, J.; Fernandes, K.J.; Abdouh, M.; Bernier, G. Modeling Late-Onset Sporadic Alzheimer’s Disease through BMI1 Deficiency. Cell Rep. 2018, 23, 2653–2666. [Google Scholar] [CrossRef] [PubMed]
- Sodersten, E.; Feyder, M.; Lerdrup, M.; Gomes, A.L.; Kryh, H.; Spigolon, G.; Caboche, J.; Fisone, G.; Hansen, K. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLoS Genet. 2014, 10, e1004574. [Google Scholar] [CrossRef]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s Disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar] [CrossRef]
- Kohler, C.; Hennig, L. Regulation of cell identity by plant Polycomb and trithorax group proteins. Curr. Opin. Genet. Dev. 2010, 20, 541–547. [Google Scholar] [CrossRef]
- Whitcomb, S.J.; Basu, A.; Allis, C.D.; Bernstein, E. Polycomb Group proteins: An evolutionary perspective. Trends Genet. 2007, 23, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; DeCamillis, M.; Zink, D.; Cheng, N.; Brock, H.W.; Paro, R. Polycomb and polyhomeotic are constituents of a multimeric protein complex in chromatin of Drosophila melanogaster. EMBO J. 1992, 11, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.; Kotarski, M.A.; Tartof, K.D. Dosage-dependent modifiers of position effect variegation in Drosophila and a mass action model that explains their effect. Genetics 1988, 120, 181–198. [Google Scholar] [PubMed]
- Schwartz, Y.B.; Pirrotta, V. Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 2008, 20, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Raible, F.; Mollaaghababa, R.; Guyon, J.R.; Wu, C.T.; Bender, W.; Kingston, R.E. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 1999, 98, 37–46. [Google Scholar] [CrossRef]
- Ng, J.; Hart, C.M.; Morgan, K.; Simon, J.A. A Drosophila ESC-E(Z) protein complex is distinct from other polycomb group complexes and contains covalently modified ESC. Mol. Cell Biol. 2000, 20, 3069–3078. [Google Scholar] [CrossRef]
- Tie, F.; Furuyama, T.; Prasad-Sinha, J.; Jane, E.; Harte, P.J. The Drosophila Polycomb Group proteins ESC and E(Z) are present in a complex containing the histone-binding protein p55 and the histone deacetylase RPD3. Development 2001, 128, 275–286. [Google Scholar]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Ketel, C.S.; Andersen, E.F.; Vargas, M.L.; Suh, J.; Strome, S.; Simon, J.A. Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Mol. Cell Biol. 2005, 25, 6857–6868. [Google Scholar] [CrossRef]
- Cooney, E.; Bi, W.; Schlesinger, A.E.; Vinson, S.; Potocki, L. Novel EED mutation in patient with Weaver syndrome. Am. J. Med. Genet. A 2017, 173, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, E.; Albuquerque, E.V.A.; Isidor, B.; Mitsuhashi, S.; Mizuguchi, T.; Miyatake, S.; Takata, A.; Miyake, N.; Boguszewski, M.C.S.; Boguszewski, C.L.; et al. Novel SUZ12 mutations in Weaver-like syndrome. Clin. Genet. 2018, 94, 461–466. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Hanks, S.; Ruark, E.; Zachariou, A.; Duarte Sdel, V.; Ramsay, E.; Snape, K.; Murray, A.; Perdeaux, E.R.; Seal, S.; et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2011, 2, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.S.; Yap, D.B.; Lewis, M.E.; Chijiwa, C.; Ramos-Arroyo, M.A.; Tkachenko, N.; Milano, V.; Fradin, M.; McKinnon, M.L.; Townsend, K.N.; et al. Weaver Syndrome-Associated EZH2 Protein Variants Show Impaired Histone Methyltransferase Function In Vitro. Hum. Mutat. 2016, 37, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, E.; Higashimoto, K.; Sakai, Y.; Numakura, C.; Okamoto, N.; Matsunaga, S.; Ryo, A.; Sato, Y.; Sanefuji, M.; Ihara, K.; et al. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum. Mutat. 2017, 38, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Trupke, J.; Ringrose, L. The quest for mammalian Polycomb response elements: Are we there yet? Chromosoma 2016, 125, 471–496. [Google Scholar] [CrossRef]
- Mohd-Sarip, A.; Venturini, F.; Chalkley, G.E.; Verrijzer, C.P. Pleiohomeotic can link polycomb to DNA and mediate transcriptional repression. Mol. Cell Biol. 2002, 22, 7473–7483. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; Koche, R.P.; Truong, T.; Zhou, V.W.; Issac, B.; Chi, A.S.; Ku, M.; Bernstein, B.E. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010, 6, e1001244. [Google Scholar] [CrossRef]
- Sing, A.; Pannell, D.; Karaiskakis, A.; Sturgeon, K.; Djabali, M.; Ellis, J.; Lipshitz, H.D.; Cordes, S.P. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell 2009, 138, 885–897. [Google Scholar] [CrossRef]
- Woo, C.J.; Kharchenko, P.V.; Daheron, L.; Park, P.J.; Kingston, R.E. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell 2010, 140, 99–110. [Google Scholar] [CrossRef]
- Schorderet, P.; Lonfat, N.; Darbellay, F.; Tschopp, P.; Gitto, S.; Soshnikova, N.; Duboule, D. A genetic approach to the recruitment of PRC2 at the HoxD locus. PLoS Genet. 2013, 9, e1003951. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.; Kroll, K.L. The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res. 2015, 359, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Lander, G.C.; Maiolica, A.; Herzog, F.; Aebersold, R.; Nogales, E. Molecular architecture of human polycomb repressive complex 2. Elife 2012, 1, e00005. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ekram, M.B.; Bakshi, A.; Kim, J. AEBP2 as a transcriptional activator and its role in cell migration. Genomics 2015, 105, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, K.; Ekram, M.B.; Roh, T.Y.; Kim, J. Aebp2 as an epigenetic regulator for neural crest cells. PLoS ONE 2011, 6, e25174. [Google Scholar] [CrossRef]
- Burgold, T.; Spreafico, F.; De Santa, F.; Totaro, M.G.; Prosperini, E.; Natoli, G.; Testa, G. The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS ONE 2008, 3, e3034. [Google Scholar] [CrossRef]
- Egan, C.M.; Nyman, U.; Skotte, J.; Streubel, G.; Turner, S.; O’Connell, D.J.; Rraklli, V.; Dolan, M.J.; Chadderton, N.; Hansen, K.; et al. CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev. Cell 2013, 26, 223–236. [Google Scholar] [CrossRef]
- Fischle, W.; Wang, Y.; Jacobs, S.A.; Kim, Y.; Allis, C.D.; Khorasanizadeh, S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003, 17, 1870–1881. [Google Scholar] [CrossRef]
- Min, J.; Zhang, Y.; Xu, R.M. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003, 17, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Vincenz, C.; Kerppola, T.K. Different polycomb group CBX family proteins associate with distinct regions of chromatin using nonhomologous protein sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 16572–16577. [Google Scholar] [CrossRef]
- Bernstein, E.; Duncan, E.M.; Masui, O.; Gil, J.; Heard, E.; Allis, C.D. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell Biol. 2006, 26, 2560–2569. [Google Scholar] [CrossRef] [PubMed]
- Sewalt, R.G.; Gunster, M.J.; van der Vlag, J.; Satijn, D.P.; Otte, A.P. C-Terminal binding protein is a transcriptional repressor that interacts with a specific class of vertebrate Polycomb proteins. Mol. Cell Biol. 1999, 19, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Melhuish, T.A.; Wotton, D. The polycomb protein Pc2 is a SUMO E3. Cell 2003, 113, 127–137. [Google Scholar] [CrossRef]
- de Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef]
- Moccia, A.; Martin, D.M. Nervous system development and disease: A focus on trithorax related proteins and chromatin remodelers. Mol. Cell Neurosci. 2018, 87, 46–54. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Martinez, A.M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA 2001, 98, 12902–12907. [Google Scholar] [CrossRef]
- Roguev, A.; Schaft, D.; Shevchenko, A.; Pijnappel, W.W.; Wilm, M.; Aasland, R.; Stewart, A.F. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001, 20, 7137–7148. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shukla, A.; Wang, X.; Chen, W.Y.; Bernstein, B.E.; Roeder, R.G. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 2011, 144, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, P.F.; Lee, J.S.; Martin-Brown, S.; Florens, L.; Washburn, M.; Shilatifard, A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell Biol. 2008, 28, 7337–7344. [Google Scholar] [CrossRef] [PubMed]
- Steward, M.M.; Lee, J.S.; O’Donovan, A.; Wyatt, M.; Bernstein, B.E.; Shilatifard, A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 2006, 13, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Hannibal, M.C.; Buckingham, K.J.; Ng, S.B.; Ming, J.E.; Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Bigham, A.W.; Tabor, H.K.; Mefford, H.C.; et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A 2011, 155, 1511–1516. [Google Scholar] [CrossRef]
- Jones, W.D.; Dafou, D.; McEntagart, M.; Woollard, W.J.; Elmslie, F.V.; Holder-Espinasse, M.; Irving, M.; Saggar, A.K.; Smithson, S.; Trembath, R.C.; et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 2012, 91, 358–364. [Google Scholar] [CrossRef]
- Yang, Y.J.; Baltus, A.E.; Mathew, R.S.; Murphy, E.A.; Evrony, G.D.; Gonzalez, D.M.; Wang, E.P.; Marshall-Walker, C.A.; Barry, B.J.; Murn, J.; et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 2012, 151, 1097–1112. [Google Scholar] [CrossRef]
- Sun, Y.M.; Greenway, D.J.; Johnson, R.; Street, M.; Belyaev, N.D.; Deuchars, J.; Bee, T.; Wilde, S.; Buckley, N.J. Distinct profiles of REST interactions with its target genes at different stages of neuronal development. Mol. Biol. Cell 2005, 16, 5630–5638. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Bartholomew, B. Regulation of ATP-dependent chromatin remodelers: Accelerators/brakes, anchors and sensors. Biochem. Soc. Trans. 2018, 46, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Sokpor, G.; Castro-Hernandez, R.; Rosenbusch, J.; Staiger, J.F.; Tuoc, T. ATP-Dependent Chromatin Remodeling During Cortical Neurogenesis. Front. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, M.D.; Aslanian, A.; Dong, M.Q.; Yates, J.R., 3rd; Emerson, B.M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol.Chem 2008, 283, 32254–32263. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Wu, J.I.; Ranish, J.A.; Wan, M.; Winslow, M.M.; Staahl, B.T.; Wu, H.; Aebersold, R.; Graef, I.A.; Crabtree, G.R. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 2007, 55, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Ishiguro, H.; Yazaki, S.; Horiuchi, Y.; Arai, M.; Niizato, K.; Iritani, S.; Itokawa, M.; Inada, T.; Iwata, N.; et al. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum. Mol. Genet. 2009, 18, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Coban Akdemir, Z.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.M.; et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef]
- Yip, D.J.; Corcoran, C.P.; Alvarez-Saavedra, M.; DeMaria, A.; Rennick, S.; Mears, A.J.; Rudnicki, M.A.; Messier, C.; Picketts, D.J. Snf2l regulates Foxg1-dependent progenitor cell expansion in the developing brain. Dev. Cell 2012, 22, 871–878. [Google Scholar] [CrossRef]
- Alvarez-Saavedra, M.; De Repentigny, Y.; Lagali, P.S.; Raghu Ram, E.V.; Yan, K.; Hashem, E.; Ivanochko, D.; Huh, M.S.; Yang, D.; Mears, A.J.; et al. Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat. Commun. 2014, 5, 4181. [Google Scholar] [CrossRef]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; de Sa, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016, 53, 190–199. [Google Scholar] [CrossRef]
- Homann, O.R.; Misura, K.; Lamas, E.; Sandrock, R.W.; Nelson, P.; McDonough, S.I.; DeLisi, L.E. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol. Psychiatry 2016, 21, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Bosman, E.A.; Penn, A.C.; Ambrose, J.C.; Kettleborough, R.; Stemple, D.L.; Steel, K.P. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 2005, 14, 3463–3476. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Kawauchi, D.; Korkel-Qu, H.; Deng, H.; Serger, E.; Sieber, L.; Lieberman, J.A.; Jimeno-Gonzalez, S.; Lambo, S.; Hanna, B.S.; et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 2017, 8, 14758. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Khan, M.A.; Bellvis, P.; Zhu, Z.; Bernhardt, O.; Herold-Mende, C.; Liu, H.K. The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell 2013, 13, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, Y.; Xian, L.; Chen, W.; Wu, H.; Gao, X. The mutation in Chd7 causes misexpression of Bmp4 and developmental defects in telencephalic midline. Am. J. Pathol. 2012, 181, 626–641. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Marie, C.; Zhao, C.; Kim, B.; Wang, J.; Deng, Y.; Clavairoly, A.; Frah, M.; Wang, H.; He, X.; et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci. 2016, 19, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; van Ravenswaaij, C.M.; Admiraal, R.; Hurst, J.A.; de Vries, B.B.; Janssen, I.M.; van der Vliet, W.A.; Huys, E.H.; de Jong, P.J.; Hamel, B.C.; et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef]
- Feng, W.; Shao, C.; Liu, H.K. Versatile Roles of the Chromatin Remodeler CHD7 during Brain Development and Disease. Front. Mol. Neurosci. 2017, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R Soc. Lond. B Biol.Sci. 2017, 372, 20160290. [Google Scholar] [CrossRef]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef]
- Lynch, M.D.; Smith, A.J.; De Gobbi, M.; Flenley, M.; Hughes, J.R.; Vernimmen, D.; Ayyub, H.; Sharpe, J.A.; Sloane-Stanley, J.A.; Sutherland, L.; et al. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J. 2012, 31, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Kirk, B.; Zeng, J.; Ma, J.; Wang, Q. Three classes of response elements for human PRC2 and MLL1/2-Trithorax complexes. Nucleic Acids Res. 2018, 46, 8848–8864. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Li, Y.; Wang, Z.; Chen, L.; Poidevin, M.; Zhang, C.; Lin, L.; Wang, F.; Bao, H.; Jiao, B.; et al. Active N(6)-Methyladenine Demethylation by DMAD Regulates Gene Expression by Coordinating with Polycomb Protein in Neurons. Mol. Cell 2018, 71, 848–857.e6. [Google Scholar] [CrossRef] [PubMed]
- Kweon, S.M.; Chen, Y.; Moon, E.; Kvederaviciute, K.; Klimasauskas, S.; Feldman, D.E. An Adversarial DNA N(6)-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell 2019, 74, 1138–1147.e6. [Google Scholar] [CrossRef]
- Yao, B.; Christian, K.M.; He, C.; Jin, P.; Ming, G.L.; Song, H. Epigenetic mechanisms in neurogenesis. Nat. Rev. Neurosci. 2016, 17, 537–549. [Google Scholar] [CrossRef]
- Kaplan, M.S.; Hinds, J.W. Neurogenesis in the adult rat: Electron microscopic analysis of light radioautographs. Science 1977, 197, 1092–1094. [Google Scholar] [CrossRef]
- Gotz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.; Stoykova, A.; Gruss, P. Differential expression of polycomb repression complex 1 (PRC1) members in the developing mouse brain reveals multiple complexes. Dev. Dyn. 2006, 235, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Suzki, N.; Tsuboi, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Koseki, H.; Vidal, M.; Gotoh, Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009, 63, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.E.; Dykhuizen, E.C. Compositional and functional diversity of canonical PRC1 complexes in mammals. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 233–245. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.A.; Phoenix, T.N.; Kokovay, E.; Lowry, N.; Elkabetz, Y.; Dimos, J.T.; Lemischka, I.R.; Studer, L.; Temple, S. Bmi-1 cooperates with Foxg1 to maintain neural stem cell self-renewal in the forebrain. Genes Dev. 2009, 23, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Xu, Y.J.; Teng, Z.Q.; Liu, C.M. Polycomb Repressive Complex 2: Emerging Roles in the Central Nervous System. Neuroscientist 2018, 24, 208–220. [Google Scholar] [CrossRef]
- Lehmann, O.J.; Sowden, J.C.; Carlsson, P.; Jordan, T.; Bhattacharya, S.S. Fox’s in development and disease. Trends Genet. 2003, 19, 339–344. [Google Scholar] [CrossRef]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef]
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty pairs of sox: Extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev. Cell 2002, 3, 167–170. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [PubMed]
- Zemke, M.; Draganova, K.; Klug, A.; Scholer, A.; Zurkirchen, L.; Gay, M.H.; Cheng, P.; Koseki, H.; Valenta, T.; Schubeler, D.; et al. Loss of Ezh2 promotes a midbrain-to-forebrain identity switch by direct gene derepression and Wnt-dependent regulation. BMC Biol. 2015, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Juan, A.H.; Wang, H.A.; Ko, K.D.; Zare, H.; Sartorelli, V. Polycomb Ezh2 controls the fate of GABAergic neurons in the embryonic cerebellum. Development 2016, 143, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.W.; Salinas, R.D.; Siu, J.J.; Kelley, K.W.; Delgado, R.N.; Paredes, M.F.; Alvarez-Buylla, A.; Oldham, M.C.; Lim, D.A. Distinct and separable roles for EZH2 in neurogenic astroglia. Elife 2014, 3, e02439. [Google Scholar] [CrossRef]
- Sun, B.; Chang, E.; Gerhartl, A.; Szele, F.G. Polycomb Protein Eed is Required for Neurogenesis and Cortical Injury Activation in the Subventricular Zone. Cereb. Cortex 2018, 28, 1369–1382. [Google Scholar] [CrossRef]
- Kassis, J.A.; Kennison, J.A.; Tamkun, J.W. Polycomb and Trithorax Group Genes in Drosophila. Genetics 2017, 206, 1699–1725. [Google Scholar] [CrossRef]
- Philippidou, P.; Dasen, J.S. Hox genes: Choreographers in neural development, architects of circuit organization. Neuron 2013, 80, 12–34. [Google Scholar] [CrossRef]
- Holland, L.Z.; Carvalho, J.E.; Escriva, H.; Laudet, V.; Schubert, M.; Shimeld, S.M.; Yu, J.K. Evolution of bilaterian central nervous systems: A single origin? Evodevo 2013, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Yaghmaeian Salmani, B.; Monedero Cobeta, I.; Rakar, J.; Bauer, S.; Curt, J.R.; Starkenberg, A.; Thor, S. Evolutionarily conserved anterior expansion of the central nervous system promoted by a common PcG-Hox program. Development 2018, 145, dev160747. [Google Scholar] [CrossRef] [PubMed]
- Curt, J.R.; Yaghmaeian Salmani, B.; Thor, S. Anterior CNS expansion driven by brain transcription factors. Elife 2019, 8, e45274. [Google Scholar] [CrossRef] [PubMed]
- Monedero Cobeta, I.; Salmani, B.Y.; Thor, S. Anterior-Posterior Gradient in Neural Stem and Daughter Cell Proliferation Governed by Spatial and Temporal Hox Control. Curr. Biol. 2017, 27, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Cao, J.L.; Hu, Y.; Yang, J.G.; Ji, Y.; Huang, J.; Zhang, Y.; Sun, D.G.; Xia, H.F.; Ma, X. The dynamics of polycomb group proteins in early embryonic nervous system in mouse and human. Int. J. Dev. Neurosci. 2013, 31, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Faust, C.; Lawson, K.A.; Schork, N.J.; Thiel, B.; Magnuson, T. The Polycomb-group gene eed is required for normal morphogenetic movements during gastrulation in the mouse embryo. Development 1998, 125, 4495–4506. [Google Scholar] [PubMed]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Miro, X.; Zhou, X.; Boretius, S.; Michaelis, T.; Kubisch, C.; Alvarez-Bolado, G.; Gruss, P. Haploinsufficiency of the murine polycomb gene Suz12 results in diverse malformations of the brain and neural tube. Dis. Model. Mech. 2009, 2, 412–418. [Google Scholar] [CrossRef]
- Satrimafitrah, P.; Barman, H.K.; Ahmad, A.; Nishitoh, H.; Nakayama, T.; Fukagawa, T.; Takami, Y. RbAp48 is essential for viability of vertebrate cells and plays a role in chromosome stability. Chromosome Res. 2016, 24, 161–173. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef]
- Bergsland, M.; Werme, M.; Malewicz, M.; Perlmann, T.; Muhr, J. The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 2006, 20, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Huang, Y.C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nowakowski, R.S.; Caviness, V.S., Jr. Mode of cell proliferation in the developing mouse neocortex. Proc. Natl. Acad. Sci. USA 1994, 91, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Chenn, A.; McConnell, S.K. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 1995, 82, 631–641. [Google Scholar] [CrossRef]
- Sun, Y.; Nadal-Vicens, M.; Misono, S.; Lin, M.Z.; Zubiaga, A.; Hua, X.; Fan, G.; Greenberg, M.E. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 2001, 104, 365–376. [Google Scholar] [CrossRef]
- Sher, F.; Rossler, R.; Brouwer, N.; Balasubramaniyan, V.; Boddeke, E.; Copray, S. Differentiation of neural stem cells into oligodendrocytes: Involvement of the polycomb group protein Ezh2. Stem Cells 2008, 26, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Koreman, E.; Sun, X.; Lu, Q.R. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol. Cell Neurosci. 2018, 87, 18–26. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.; Mao, M.; et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 2013, 152, 248–261. [Google Scholar] [CrossRef]
- Di Meglio, T.; Kratochwil, C.F.; Vilain, N.; Loche, A.; Vitobello, A.; Yonehara, K.; Hrycaj, S.M.; Roska, B.; Peters, A.H.; Eichmann, A.; et al. Ezh2 orchestrates topographic migration and connectivity of mouse precerebellar neurons. Science 2013, 339, 204–207. [Google Scholar] [CrossRef]
- Kratochwil, C.F.; Maheshwari, U.; Rijli, F.M. The Long Journey of Pontine Nuclei Neurons: From Rhombic Lip to Cortico-Ponto-Cerebellar Circuitry. Front. Neural Circuits 2017, 11, 33. [Google Scholar] [CrossRef]
- Nitarska, J.; Smith, J.G.; Sherlock, W.T.; Hillege, M.M.; Nott, A.; Barshop, W.D.; Vashisht, A.A.; Wohlschlegel, J.A.; Mitter, R.; Riccio, A. A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development. Cell Rep. 2016, 17, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Nitarska, J.; Veenvliet, J.V.; Schacke, S.; Derijck, A.A.; Sirko, P.; Muchardt, C.; Pasterkamp, R.J.; Smidt, M.P.; Riccio, A. S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proc. Natl. Acad. Sci. USA 2013, 110, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Watson, P.M.; Robinson, J.D.; Crepaldi, L.; Riccio, A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 2008, 455, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.A.; Stasiv, Y.; Benraiss, A.; Chmielnicki, E.; Grinberg, A.; Westphal, H.; Goldman, S.A.; Enikolopov, G. Nitric oxide negatively regulates mammalian adult neurogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 9566–9571. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yoshida, S.; Yano, M.; Hanaoka, F. Roles of endogenous nitric oxide in cerebellar cortical development in slice cultures. Neuroreport 1994, 5, 2049–2052. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e48. [Google Scholar] [CrossRef] [PubMed]
- Stenzel-Poore, M.P.; Stevens, S.L.; Xiong, Z.; Lessov, N.S.; Harrington, C.A.; Mori, M.; Meller, R.; Rosenzweig, H.L.; Tobar, E.; Shaw, T.E. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: Similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003, 362, 1028–1037. [Google Scholar] [CrossRef]
- Stapels, M.; Piper, C.; Yang, T.; Li, M.; Stowell, C.; Xiong, Z.G.; Saugstad, J.; Simon, R.P.; Geromanos, S.; Langridge, J.; et al. Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci. Signal. 2010, 3, ra15. [Google Scholar] [CrossRef]
- Wei, L.; Yu, S.P.; Gottron, F.; Snider, B.J.; Zipfel, G.J.; Choi, D.W. Potassium channel blockers attenuate hypoxia- and ischemia-induced neuronal death in vitro and in vivo. Stroke 2003, 34, 1281–1286. [Google Scholar] [CrossRef]
- Iaci, J.F.; Parry, T.J.; Huang, Z.; Finklestein, S.P.; Ren, J.; Barrile, D.K.; Davenport, M.D.; Wu, R.; Blight, A.R.; Caggiano, A.O. Dalfampridine improves sensorimotor function in rats with chronic deficits after middle cerebral artery occlusion. Stroke 2013, 44, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.; Cortes, M.; Rykman, A.; Hill, J.; Karuppagounder, S.; Edwards, D.; Ratan, R.R. The epigenetics of stroke recovery and rehabilitation: From polycomb to histone deacetylases. Neurotherapeutics 2013, 10, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.K.; Tabor, C.W.; Tabor, H. Polyamine deficiency leads to accumulation of reactive oxygen species in a spe2Delta mutant of Saccharomyces cerevisiae. Yeast 2006, 23, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Soda, K.; Dobashi, Y.; Kano, Y.; Tsujinaka, S.; Konishi, F. Polyamine-rich food decreases age-associated pathology and mortality in aged mice. Exp. Gerontol. 2009, 44, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Chatoo, W.; Abdouh, M.; David, J.; Champagne, M.P.; Ferreira, J.; Rodier, F.; Bernier, G. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J. Neurosci. 2009, 29, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.H.; Poon, P.C.; Glatt-Deeley, H.; Abrams, J.M.; Helfand, S.L. Neuronal expression of p53 dominant-negative proteins in adult Drosophila melanogaster extends life span. Curr. Biol. 2005, 15, 2063–2068. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef]
- Matheu, A.; Maraver, A.; Klatt, P.; Flores, I.; Garcia-Cao, I.; Borras, C.; Flores, J.M.; Vina, J.; Blasco, M.A.; Serrano, M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007, 448, 375–379. [Google Scholar] [CrossRef]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Martin, J.B.; Gusella, J.F. Huntington’s disease. Pathogenesis and management. N. Engl. J. Med. 1986, 315, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Woda, J.M.; Calzonetti, T.; Hilditch-Maguire, P.; Duyao, M.P.; Conlon, R.A.; MacDonald, M.E. Inactivation of the Huntington’s disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev. Biol. 2005, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Woda, J.M.; Song, J.J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Ferrari, F.; Mendenhall, E.M.; Zhang, Y.; Erdin, S.; Vijayvargia, R.; Vallabh, S.M.; Solomos, N.; Manavalan, P.; Ragavendran, A.; et al. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 2015, 24, 2442–2457. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tsuji, J.; Labadorf, A.; Roussos, P.; Chen, J.F.; Myers, R.H.; Akbarian, S.; Weng, Z. The Role of H3K4me3 in Transcriptional Regulation Is Altered in Huntington’s Disease. PLoS ONE 2015, 10, e0144398. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R. Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol. Dis. 2012, 46, 245–254. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Bermudez, J. Alzheimer’s disease: Critical notes on the history of a medical concept. Arch. Med. Res. 2012, 43, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. The genetics of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 107, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Kerimoglu, C.; Sakib, M.S.; Jain, G.; Benito, E.; Burkhardt, S.; Capece, V.; Kaurani, L.; Halder, R.; Agis-Balboa, R.C.; Stilling, R.; et al. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep. 2017, 20, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.C.; Zhang, P.; Wurster, A.L.; Precht, P.; Mughal, M.R.; Wood, W.H., 3rd; Zhang, Y.; Becker, K.G.; Mattson, M.P.; Pazin, M.J. CHD5, a brain-specific paralog of Mi2 chromatin remodeling enzymes, regulates expression of neuronal genes. PLoS ONE 2011, 6, e24515. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Gotoh, T.; Kok, M.; White, P.S.; Brodeur, G.M. CHD5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene 2003, 22, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, H.J. The basal ganglia and motor control. Neural. Plast. 2003, 10, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.J.; Kumar, V.B.; Pandey, N.; Panneton, W.M.; Gan, Q.; Franko, M.W.; O’Dell, M.; Li, S.W.; Pan, Y.; Chung, H.D.; et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008, 115, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Kruger, R.; Muller, T.; Reichmann, H.; et al. alpha-Synuclein in Parkinson’s disease: Causal or bystander? J. Neural Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef] [PubMed]
- Birkmayer, W.; Hornykiewicz, O. The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. Parkinsonism Relat. Disord. 1998, 4, 59–60. [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. The L-dihydroxyphenylalanine (L-DOPA) effect in Parkinson’s syndrome in man: On the pathogenesis and treatment of Parkinson akinesis. Arch. Psychiatr. Nervenkr. Z Gesamte. Neurol. Psychiatr. 1962, 203, 560–574. [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien. Klin. Wochenschr. 1961, 73, 787–788. [Google Scholar]
- Aubert, I.; Guigoni, C.; Håkansson, K.; Li, Q.; Dovero, S.; Barthe, N.; Bioulac, B.H.; Gross, C.E.; Fisone, G.; Bloch, B. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann. Neurol. 2005, 57, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Tanimura, A.; Graves, S.M.; Shen, W.; Surmeier, D.J. Striatal synapses, circuits, and Parkinson’s disease. Curr. Opin. Neurobiol. 2018, 48, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Srinageshwar, B.; Maiti, P.; Dunbar, G.L.; Rossignol, J. Role of Epigenetics in Stem Cell Proliferation and Differentiation: Implications for Treating Neurodegenerative Diseases. Int. J. Mol. Sci. 2016, 17, 199. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Spencer, B.; Michael, S.; Castello, N.A.; Agazaryan, A.A.; Davis, J.L.; Muller, F.J.; Loring, J.F.; Masliah, E.; LaFerla, F.M. Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.T.; Rossignol, J.; Dunbar, G.L. Use of Genetically Altered Stem Cells for the Treatment of Huntington’s Disease. Brain Sci. 2014, 4, 202–219. [Google Scholar] [CrossRef] [PubMed]
- d’Anglemont de Tassigny, X.; Pascual, A.; Lopez-Barneo, J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front. Neuroanat. 2015, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Collas, P.; Noer, A.; Sorensen, A.L. Epigenetic Basis for the Differentiation Potential of Mesenchymal and Embryonic Stem Cells. Transfus Med. Hemother. 2008, 35, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Noer, A.; Lindeman, L.C.; Collas, P. Histone H3 modifications associated with differentiation and long-term culture of mesenchymal adipose stem cells. Stem Cells Dev. 2009, 18, 725–736. [Google Scholar] [CrossRef]
- McGinnis, W.; Krumlauf, R. Homeobox genes and axial patterning. Cell 1992, 68, 283–302. [Google Scholar] [CrossRef]
- Abreu, C.M.; Gama, L.; Krasemann, S.; Chesnut, M.; Odwin-Dacosta, S.; Hogberg, H.T.; Hartung, T.; Pamies, D. Microglia Increase Inflammatory Responses in iPSC-Derived Human BrainSpheres. Front. Microbiol. 2018, 9, 2766. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.Y.; Lombroso, A.P.; Hwang, S.M.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 2017, 21, 383–398.e7. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.A.; Reumann, D.; Bian, S.; Levi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Stem Cell Models of Human Brain Development. Cell Stem Cell 2016, 18, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Cederquist, G.Y.; Asciolla, J.J.; Tchieu, J.; Walsh, R.M.; Cornacchia, D.; Resh, M.D.; Studer, L. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 2019, 37, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Wells, J.M. Organoids by design. Science 2019, 364, 956–959. [Google Scholar] [CrossRef]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Scheijen, B.; Voncken, J.W.; Kieboom, K.; Berns, A.; van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Wagener, N.; Macher-Goeppinger, S.; Pritsch, M.; Husing, J.; Hoppe-Seyler, K.; Schirmacher, P.; Pfitzenmaier, J.; Haferkamp, A.; Hoppe-Seyler, F.; Hohenfellner, M. Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer 2010, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Takawa, M.; Masuda, K.; Kunizaki, M.; Daigo, Y.; Takagi, K.; Iwai, Y.; Cho, H.S.; Toyokawa, G.; Yamane, Y.; Maejima, K.; et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011, 102, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Uchimaru, K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 2017, 29, 375–381. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuehner, J.N.; Yao, B. The Dynamic Partnership of Polycomb and Trithorax in Brain Development and Diseases. Epigenomes 2019, 3, 17. https://doi.org/10.3390/epigenomes3030017
Kuehner JN, Yao B. The Dynamic Partnership of Polycomb and Trithorax in Brain Development and Diseases. Epigenomes. 2019; 3(3):17. https://doi.org/10.3390/epigenomes3030017
Chicago/Turabian StyleKuehner, Janise N., and Bing Yao. 2019. "The Dynamic Partnership of Polycomb and Trithorax in Brain Development and Diseases" Epigenomes 3, no. 3: 17. https://doi.org/10.3390/epigenomes3030017
APA StyleKuehner, J. N., & Yao, B. (2019). The Dynamic Partnership of Polycomb and Trithorax in Brain Development and Diseases. Epigenomes, 3(3), 17. https://doi.org/10.3390/epigenomes3030017