LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells


 and
and 
Abstract
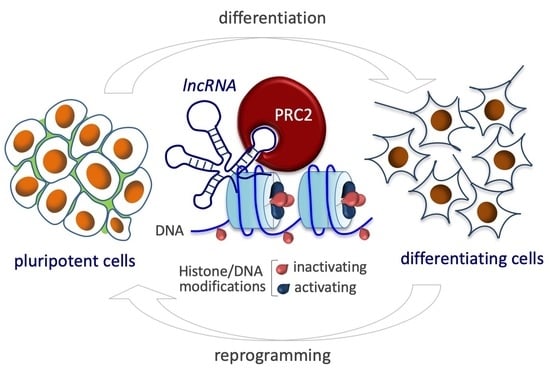
1. Introduction
2. Nuclear-Retained LncRNAs and Their Functions
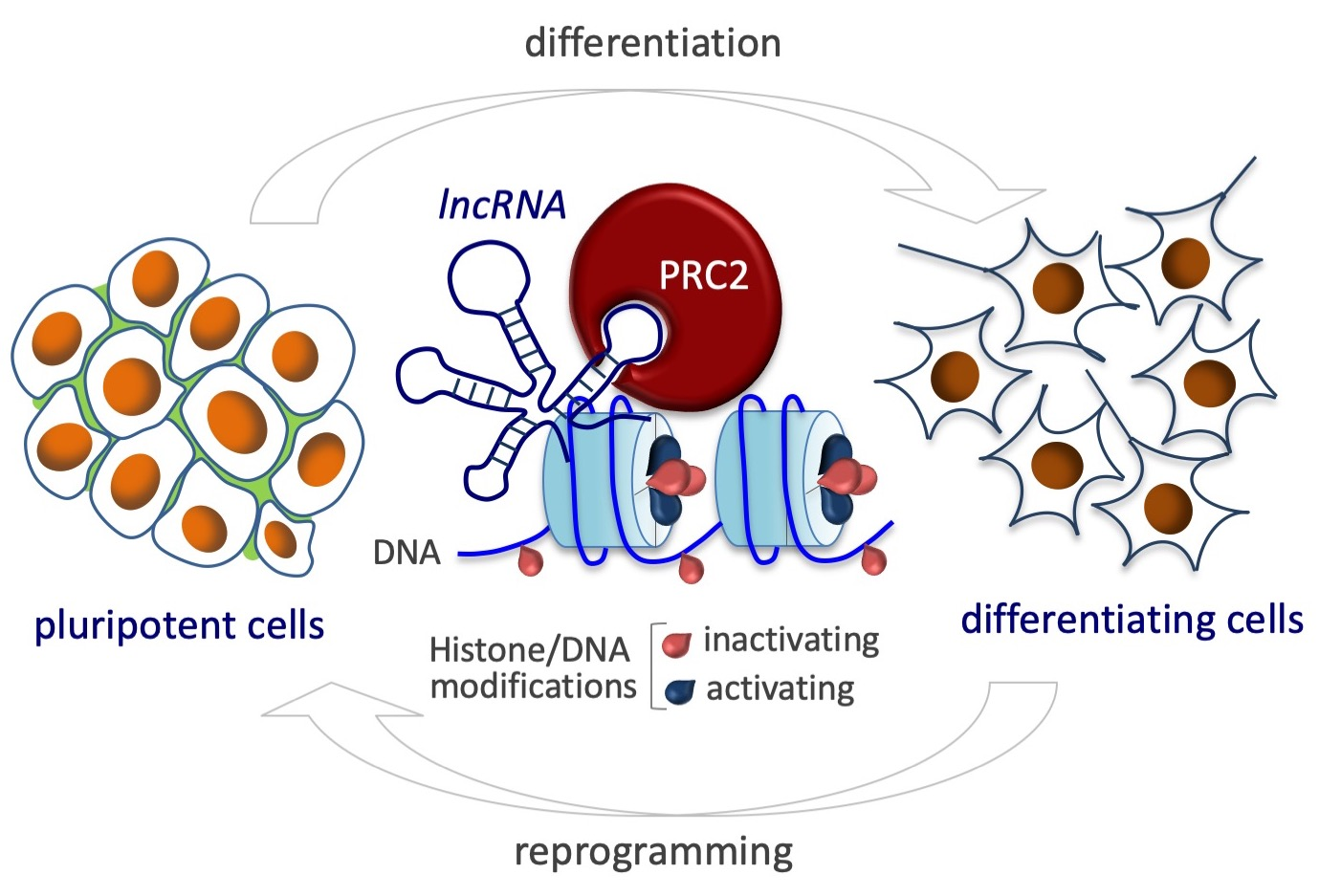
3. Polycomb Repressive Complex 2 in Embryonic Stem Cells
4. LncRNA-Directed Chromatin Regulation via PRC2 in Cell Fate
4.1. X Inactive-Specific Transcript (Xist)
4.2. Maternally Expressed 3 (Meg3)
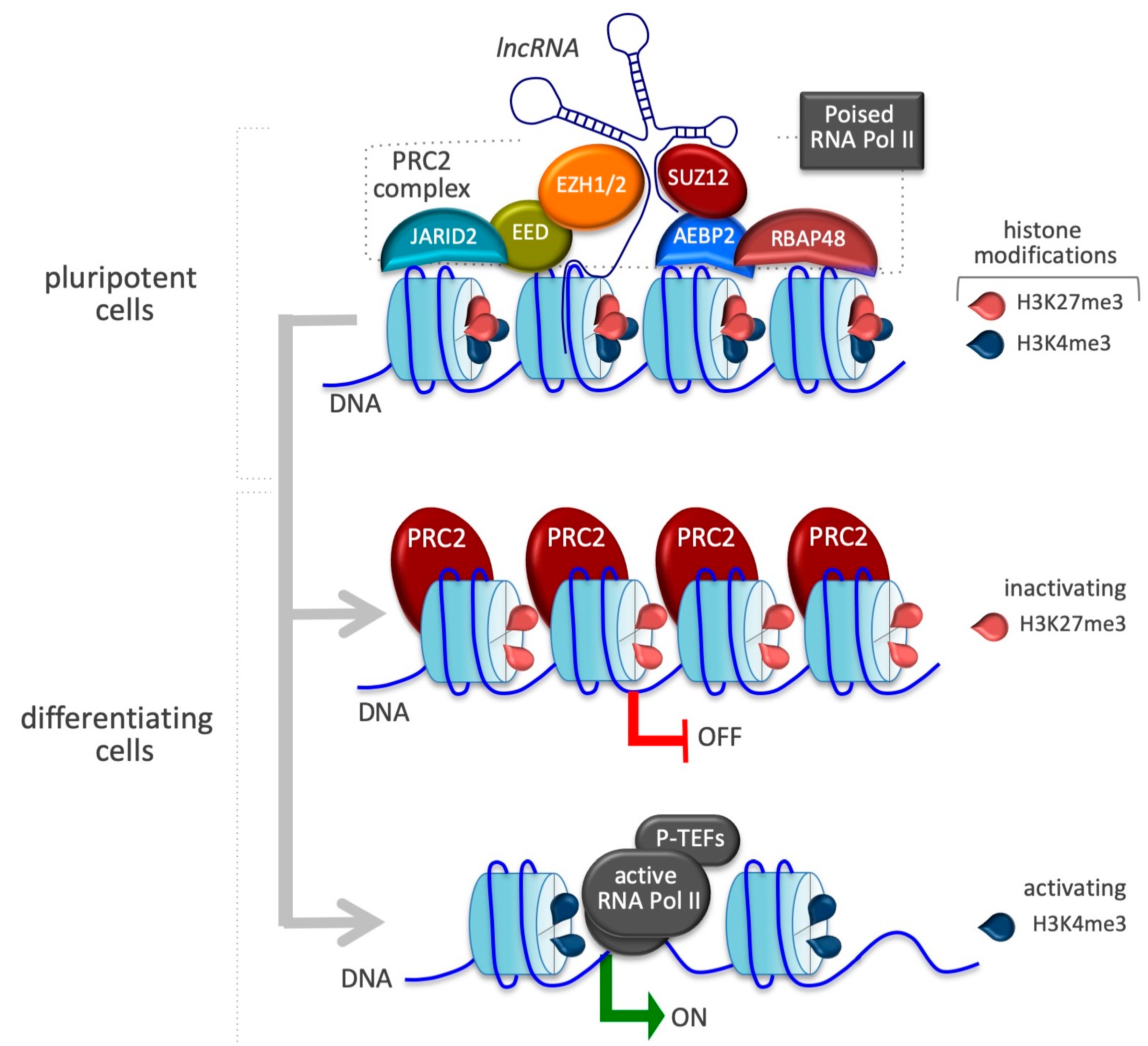
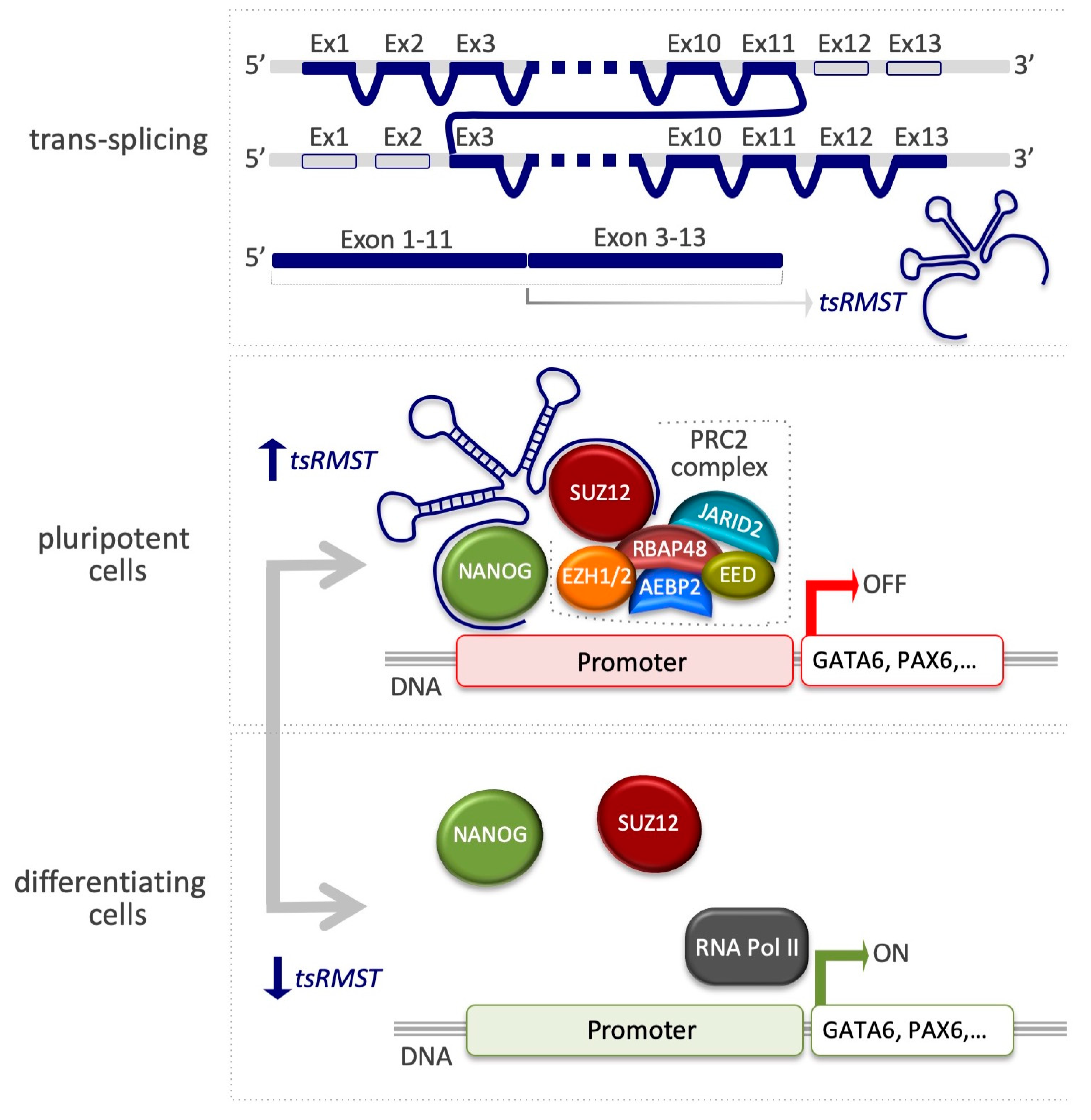
4.3. Trans-Spliced Rhabdomyosarcoma 2 Associated Transcript (tsRMST)
4.4. Long Intergenic Noncoding HOXA1 (Linc-HOXA1)
4.5. The Transcribed Ultraconserved LncRNA T-UCstem1
4.6. Heart-Associated LncRNA Braveheart (Bvht)
5. State of the Art and Future Perspectives
5.1. Current and Future Methodologies
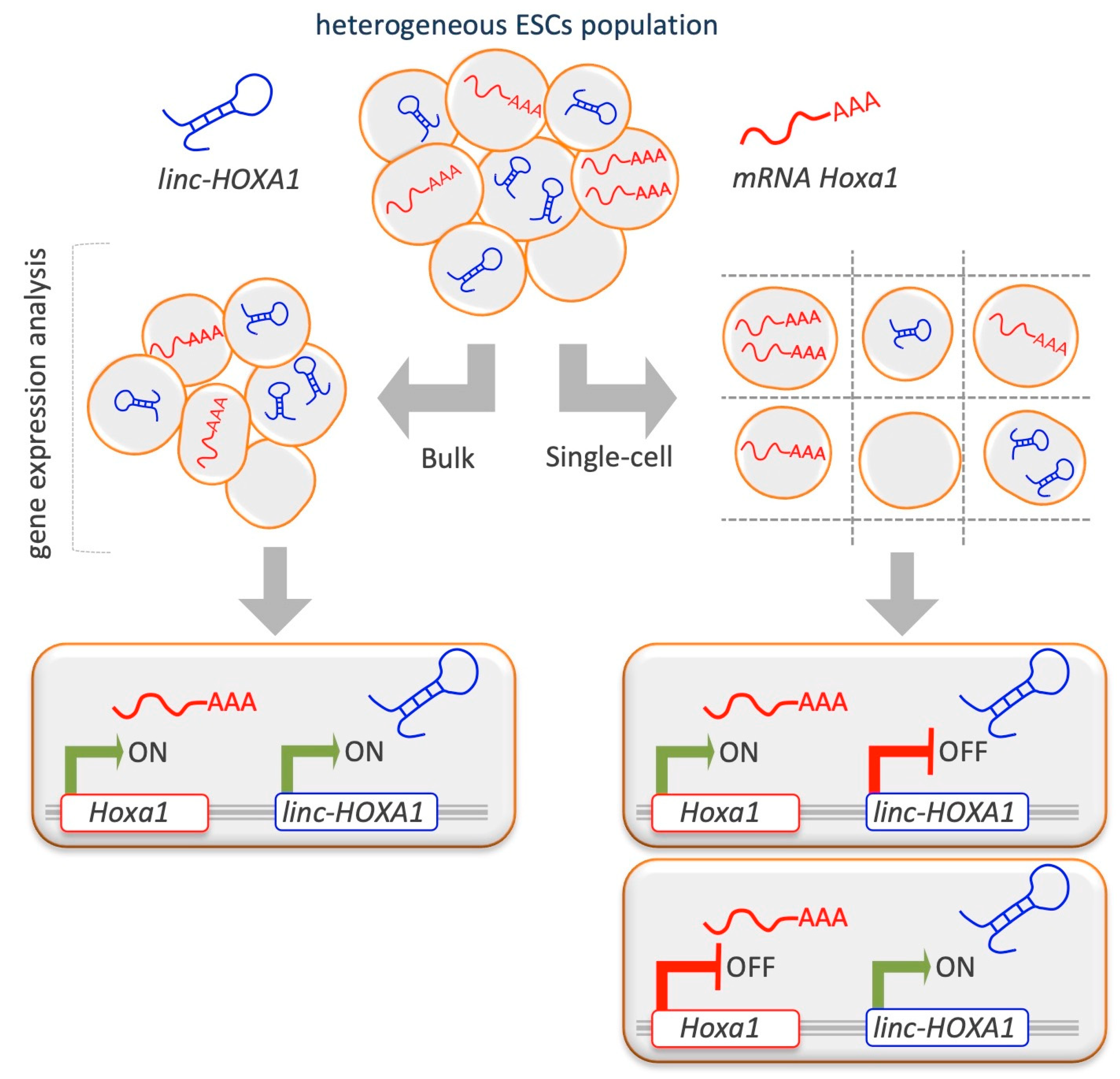
5.2. The Advent of Single-Cell Technologies: Limits and Potential
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. Microrna-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Scholer, H.R. Regulatory networks in embryo-derived pluripotent stem cells. Nat. Rev. Mol. Cell Biol. 2005, 6, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode v7 catalog of human long noncoding rnas: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human hox loci by noncoding rnas. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding rnas. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Zheng, H.; Li, J.; Liu, D.; Li, H.; Samudrala, R.; Yu, J.; Wong, G.K. Mouse transcriptome: Neutral evolution of ‘non-coding’ complementary dnas. Nature 2004, 431, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Ballarino, M. Long noncoding rna regulation of pluripotency. Stem Cells Int. 2016, 2016, 1797692. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Johnson, R.; Stanton, L.W. Human long non-coding rnas promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. Embo J. 2012, 31, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding rnas. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Wight, M.; Werner, A. The functions of natural antisense transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Shiekhattar, R. Long noncoding rnas usher in a new era in the biology of enhancers. Cell 2013, 154, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Fiorenzano, A.; Pascale, E.; Gagliardi, M.; Terreri, S.; Papa, M.; Andolfi, G.; Galasso, M.; Tagliazucchi, G.M.; Taccioli, C.; Patriarca, E.J.; et al. An ultraconserved element containing lncrna preserves transcriptional dynamics and maintains esc self-renewal. Stem Cell Rep. 2018, 10, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding rnas. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Dostie, J. Reciprocal regulation of chromatin state and architecture by hotairm1 contributes to temporal collinear hoxa gene activation. Nucleic Acids Res. 2017, 45, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, Z.; Xu, X.; Liu, G.; Song, X.; Wang, R.; Sui, X.; Liu, T.; Chang, X.; Huang, D. As1dhrs4, a head-to-head natural antisense transcript, silences the dhrs4 gene cluster in cis and trans. Proc. Natl. Acad. Sci. USA 2012, 109, 14110–14115. [Google Scholar] [CrossRef]
- Sarropoulos, I.; Marin, R.; Cardoso-Moreira, M.; Kaessmann, H. Developmental dynamics of lncrnas across mammalian organs and species. Nature 2019, 571, 510–514. [Google Scholar] [CrossRef]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncrnas at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Ulitsky, I.; Bartel, D.P. Lincrnas: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding rnas in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding rna-ror modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Fico, A.; Fiorenzano, A.; Pascale, E.; Patriarca, E.J.; Minchiotti, G. Long non-coding rna in stem cell pluripotency and lineage commitment: Functions and evolutionary conservation. Cell. Mol. Life Sci. 2019, 76, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Scarola, M.; Comisso, E.; Pascolo, R.; Chiaradia, R.; Marion, R.M.; Schneider, C.; Blasco, M.A.; Schoeftner, S.; Benetti, R. Epigenetic silencing of oct4 by a complex containing suv39h1 and oct4 pseudogene lncrna. Nat. Commun. 2015, 6, 7631. [Google Scholar] [CrossRef]
- Pal, D.; Rao, M.R.S. Long noncoding rnas in pluripotency of stem cells and cell fate specification. Adv. Exp. Med. Biol. 2017, 1008, 223–252. [Google Scholar]
- Amandio, A.R.; Necsulea, A.; Joye, E.; Mascrez, B.; Duboule, D. Hotair is dispensible for mouse development. PLoS Genet. 2016, 12, e1006232. [Google Scholar] [CrossRef]
- Ip, J.Y.; Sone, M.; Nashiki, C.; Pan, Q.; Kitaichi, K.; Yanaka, K.; Abe, T.; Takao, K.; Miyakawa, T.; Blencowe, B.J.; et al. Gomafu lncrna knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci. Rep. 2016, 6, 27204. [Google Scholar] [CrossRef]
- Bell, C.C.; Amaral, P.P.; Kalsbeek, A.; Magor, G.W.; Gillinder, K.R.; Tangermann, P.; di Lisio, L.; Cheetham, S.W.; Gruhl, F.; Frith, J.; et al. The evx1/evx1as gene locus regulates anterior-posterior patterning during gastrulation. Sci. Rep. 2016, 6, 26657. [Google Scholar] [CrossRef]
- Han, X.; Luo, S.; Peng, G.; Lu, J.Y.; Cui, G.; Liu, L.; Yan, P.; Yin, Y.; Liu, W.; Wang, R.; et al. Mouse knockout models reveal largely dispensable but context-dependent functions of lncrnas during development. J. Mol. Cell Biol. 2018, 10, 175–178. [Google Scholar] [CrossRef]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Goff, L.A.; Lodato, S.; Bonev, B.; Groff, A.F.; Gerhardinger, C.; Sanchez-Gomez, D.B.; Hacisuleyman, E.; Li, E.; Spence, M.; et al. Multiple knockout mouse models reveal lincrnas are required for life and brain development. eLife 2013, 2, e01749. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear long noncoding rnas: Key regulators of gene expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding rnas: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ye, B.; Yang, L.; Zhu, X.; Huang, G.; Zhu, P.; Du, Y.; Wu, J.; Qin, X.; Chen, R.; et al. Long noncoding rna lnckdm2b is required for ilc3 maintenance by initiation of zfp292 expression. Nat. Immunol. 2017, 18, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. Meg3 long noncoding rna regulates the tgf-beta pathway genes through formation of rna-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, I.; Munita, R.; Agirre, E.; Dittmer, T.A.; Gysling, K.; Misteli, T.; Luco, R.F. A lncrna regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat. Struct. Mol. Biol. 2015, 22, 370–376. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, A.; Ho, T.T.; Zhang, Z.; Zhou, N.; Ding, X.; Zhang, X.; Xu, M.; Mo, Y.Y. Linc-ror promotes c-myc expression through hnrnp i and auf1. Nucleic Acids Res. 2016, 44, 3059–3069. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding rna hotair in protein ubiquitination. Nat. Commun. 2013, 4, 2939. [Google Scholar] [CrossRef]
- Puvvula, P.K.; Desetty, R.D.; Pineau, P.; Marchio, A.; Moon, A.; Dejean, A.; Bischof, O. Long noncoding rna panda and scaffold-attachment-factor safa control senescence entry and exit. Nat. Commun. 2014, 5, 5323. [Google Scholar] [CrossRef]
- Chalei, V.; Sansom, S.N.; Kong, L.; Lee, S.; Montiel, J.F.; Vance, K.W.; Ponting, C.P. The long non-coding rna dali is an epigenetic regulator of neural differentiation. eLife 2014, 3, e04530. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.S.; Ruthenburg, A.J. Nuclear fractionation reveals thousands of chromatin-tethered noncoding rnas adjacent to active genes. Cell Rep. 2015, 12, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated rnas by rip-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Tanigawa, A.; Naganuma, T.; Ohkawa, Y.; Souquere, S.; Pierron, G.; Hirose, T. Swi/snf chromatin-remodeling complexes function in noncoding rna-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4304–4309. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef]
- Lewis, E.B. A gene complex controlling segmentation in drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Cavalli, G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 2009, 136, 3531–3542. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The polycomb complex prc2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Chan, C.S.; Rastelli, L.; Pirrotta, V. A polycomb response element in the ubx gene that determines an epigenetically inherited state of repression. EMBO J. 1994, 13, 2553–2564. [Google Scholar] [CrossRef]
- Muller, J.; Kassis, J.A. Polycomb response elements and targeting of polycomb group proteins in drosophila. Curr. Opin. Genet. Dev. 2006, 16, 476–484. [Google Scholar] [CrossRef]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of h2a restrains poised rna polymerase ii at bivalent genes in mouse es cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Isono, K.; Sharif, J.; Ohara, O.; Toyoda, T.; Ito, T.; Eskeland, R.; Bickmore, W.A.; et al. Histone h2a mono-ubiquitination is a crucial step to mediate prc1-dependent repression of developmental genes to maintain es cell identity. PLoS Genet. 2012, 8, e1002774. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone h3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of zeste/esc complexes have a histone h3 methyltransferase activity that marks chromosomal polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef]
- Muller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a drosophila polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The polycomb-group gene ezh2 is required for early mouse development. Mol. Cell. Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Yee, D.; Magnuson, T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 2008, 26, 1496–1505. [Google Scholar] [CrossRef]
- Landeira, D.; Sauer, S.; Poot, R.; Dvorkina, M.; Mazzarella, L.; Jorgensen, H.F.; Pereira, C.F.; Leleu, M.; Piccolo, F.M.; Spivakov, M.; et al. Jarid2 is a prc2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of prc1 and rna polymerase ii to developmental regulators. Nat. Cell Biol. 2010, 12, 618–624. [Google Scholar] [CrossRef]
- Shen, X.; Kim, W.; Fujiwara, Y.; Simon, M.D.; Liu, Y.; Mysliwiec, M.R.; Yuan, G.C.; Lee, Y.; Orkin, S.H. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 2009, 139, 1303–1314. [Google Scholar] [CrossRef]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. Jarid2 regulates binding of the polycomb repressive complex 2 to target genes in es cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef]
- Zhang, Z.; Jones, A.; Sun, C.W.; Li, C.; Chang, C.W.; Joo, H.Y.; Dai, Q.; Mysliwiec, M.R.; Wu, L.C.; Guo, Y.; et al. Prc2 complexes with jarid2, mtf2, and esprc2p48 in es cells to modulate es cell pluripotency and somatic cell reprogramming. Stem Cells 2011, 29, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kasinath, V.; Faini, M.; Poepsel, S.; Reif, D.; Feng, X.A.; Stjepanovic, G.; Aebersold, R.; Nogales, E. Structures of human prc2 with its cofactors aebp2 and jarid2. Science 2018, 359, 940–944. [Google Scholar] [CrossRef]
- Landeira, D.; Bagci, H.; Malinowski, A.R.; Brown, K.E.; Soza-Ried, J.; Feytout, A.; Webster, Z.; Ndjetehe, E.; Cantone, I.; Asenjo, H.G.; et al. Jarid2 coordinates nanog expression and pcp/wnt signaling required for efficient esc differentiation and early embryo development. Cell Rep. 2015, 12, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/jumonji coordinates control of prc2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-genome analysis of histone h3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Aloia, L.; Di Stefano, B.; Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 2013, 140, 2525–2534. [Google Scholar] [CrossRef]
- Flynn, R.A.; Chang, H.Y. Long noncoding rnas in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding rna as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Epigenetic regulation by long noncoding rnas. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The xist lncrna exploits three-dimensional genome architecture to spread across the x chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Pinter, S.F.; Fang, R.; Sarma, K.; Rutenberg-Schoenberg, M.; Bowman, S.K.; Kesner, B.A.; Maier, V.K.; Kingston, R.E.; Lee, J.T. High-resolution xist binding maps reveal two-step spreading during x-chromosome inactivation. Nature 2013, 504, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Mak, W.; Nesterova, T.B.; de Napoles, M.; Appanah, R.; Yamanaka, S.; Otte, A.P.; Brockdorff, N. Reactivation of the paternal x chromosome in early mouse embryos. Science 2004, 303, 666–669. [Google Scholar] [CrossRef] [PubMed]
- da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 is implicated in the initial xist-induced targeting of prc2 to the inactive x chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef]
- Pinter, S.F.; Sadreyev, R.I.; Yildirim, E.; Jeon, Y.; Ohsumi, T.K.; Borowsky, M.; Lee, J.T. Spreading of x chromosome inactivation via a hierarchy of defined polycomb stations. Genome Res. 2012, 22, 1864–1876. [Google Scholar] [CrossRef]
- Cerase, A.; Smeets, D.; Tang, Y.A.; Gdula, M.; Kraus, F.; Spivakov, M.; Moindrot, B.; Leleu, M.; Tattermusch, A.; Demmerle, J.; et al. Spatial separation of xist rna and polycomb proteins revealed by superresolution microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 2235–2240. [Google Scholar] [CrossRef]
- Tian, D.; Sun, S.; Lee, J.T. The long noncoding rna, jpx, is a molecular switch for x chromosome inactivation. Cell 2010, 143, 390–403. [Google Scholar] [CrossRef]
- Sun, S.; Del Rosario, B.C.; Szanto, A.; Ogawa, Y.; Jeon, Y.; Lee, J.T. Jpx rna activates xist by evicting ctcf. Cell 2013, 153, 1537–1551. [Google Scholar] [CrossRef]
- Saldana-Meyer, R.; Gonzalez-Buendia, E.; Guerrero, G.; Narendra, V.; Bonasio, R.; Recillas-Targa, F.; Reinberg, D. Ctcf regulates the human p53 gene through direct interaction with its natural antisense transcript, wrap53. Genes Dev. 2014, 28, 723–734. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat rna to the mouse x chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of xist rna. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous rna binding by polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Cifuentes-Rojas, C.; Ergun, A.; Del Rosario, A.; Jeon, Y.; White, F.; Sadreyev, R.; Lee, J.T. Atrx directs binding of prc2 to xist rna and polycomb targets. Cell 2014, 159, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. Pcgf3/5-prc1 initiates polycomb recruitment in x chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.G.; Meisig, J.; Nakamura, T.; Okamoto, I.; Sieber, A.; Picard, C.; Borensztein, M.; Saitou, M.; Bluthgen, N.; Heard, E. The two active x chromosomes in female escs block exit from the pluripotent state by modulating the esc signaling network. Cell Stem Cell 2014, 14, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Anguera, M.C.; Sadreyev, R.; Zhang, Z.; Szanto, A.; Payer, B.; Sheridan, S.D.; Kwok, S.; Haggarty, S.J.; Sur, M.; Alvarez, J.; et al. Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 2012, 11, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Payer, B.; Rosenberg, M.; Yamaji, M.; Yabuta, Y.; Koyanagi-Aoi, M.; Hayashi, K.; Yamanaka, S.; Saitou, M.; Lee, J.T. Tsix rna and the germline factor, prdm14, link x reactivation and stem cell reprogramming. Mol. Cell 2013, 52, 805–818. [Google Scholar] [CrossRef]
- Tchieu, J.; Kuoy, E.; Chin, M.H.; Trinh, H.; Patterson, M.; Sherman, S.P.; Aimiuwu, O.; Lindgren, A.; Hakimian, S.; Zack, J.A.; et al. Female human ipscs retain an inactive x chromosome. Cell Stem Cell 2010, 7, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, C.; Thakur, N.; Pandey, R.R. The length of the transcript encoded from the kcnq1ot1 antisense promoter determines the degree of silencing. EMBO J. 2006, 25, 2096–2106. [Google Scholar] [CrossRef]
- Lewis, A.; Mitsuya, K.; Umlauf, D.; Smith, P.; Dean, W.; Walter, J.; Higgins, M.; Feil, R.; Reik, W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 2004, 36, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding rna mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Son, J.; Shen, S.S.; Reinberg, D.; Bonasio, R. Prc2 binds active promoters and contacts nascent rnas in embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Bonasio, R.; Saldana-Meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between jarid2 and noncoding rnas regulate prc2 recruitment to chromatin. Mol. Cell 2014, 53, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Yu, C.Y.; Chuang, C.Y.; Hsiao, M.; Kao, C.F.; Kuo, H.C.; Chuang, T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Zhao, G.N.; Chen, X.F.; Hao, D.L.; Zhao, X.; Lv, X.; Liu, D.P. The long noncoding rna gm15055 represses hoxa gene expression by recruiting prc2 to the gene cluster. Nucleic Acids Res. 2016, 44, 2613–2627. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a long noncoding rna required for cardiovascular lineage commitment. Cell 2013, 152, 570–583. [Google Scholar] [CrossRef]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. Meg3 imprinted gene contribution in tumorigenesis. Int. J. Cancer 2011, 129, 773–779. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Klibanski, A. Meg3 noncoding rna: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef]
- da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian dlk1-dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef]
- Takahashi, N.; Okamoto, A.; Kobayashi, R.; Shirai, M.; Obata, Y.; Ogawa, H.; Sotomaru, Y.; Kono, T. Deletion of gtl2, imprinted non-coding rna, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 2009, 18, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.P.; Youngson, N.; Takada, S.; Seitz, H.; Reik, W.; Paulsen, M.; Cavaille, J.; Ferguson-Smith, A.C. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the dlk1-gtl2 imprinted cluster on mouse chromosome 12. Nat. Genet. 2003, 35, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qf1 in mouse induced pluripotent stem cells. Nature 2010, 465, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Markoulaki, S.; Hanna, J.H.; Faddah, D.A.; Buganim, Y.; Kim, J.; Ganz, K.; Steine, E.J.; Cassady, J.P.; Creyghton, M.P.; et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell 2011, 9, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Sherpa, C.; Rausch, J.W.; Le Grice, S.F. Structural characterization of maternally expressed gene 3 rna reveals conserved motifs and potential sites of interaction with polycomb repressive complex 2. Nucleic Acids Res. 2018, 46, 10432–10447. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincrnas in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef]
- Sanli, I.; Lalevee, S.; Cammisa, M.; Perrin, A.; Rage, F.; Lleres, D.; Riccio, A.; Bertrand, E.; Feil, R. Meg3 non-coding rna expression controls imprinting by preventing transcriptional upregulation in cis. Cell Rep. 2018, 23, 337–348. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Maniatis, T.; Tasic, B. Alternative pre-mrna splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef]
- Black, D.L.; Grabowski, P.J. Alternative pre-mrna splicing and neuronal function. Prog. Mol. Subcell. Biol. 2003, 31, 187–216. [Google Scholar] [PubMed]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Kryukov, K.; Clemente, J.C.; Komiyama, T.; Suzuki, Y.; Imanishi, T.; Ikeo, K.; Gojobori, T. The evolutionary relationship between gene duplication and alternative splicing. Gene 2008, 427, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Aigaki, T. Alternative trans-splicing: A novel mode of pre-mrna processing. Biol. Cell 2006, 98, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Gingeras, T.R. Implications of chimaeric non-co-linear transcripts. Nature 2009, 461, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Shepelev, V.; Fedorov, A. Bioinformatic analysis of exon repetition, exon scrambling and trans-splicing in humans. Bioinformatics 2006, 22, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, L.; Jiang, H.; Wang, W. Short homologous sequences are strongly associated with the generation of chimeric rnas in eukaryotes. J. Mol. Evol. 2009, 68, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Herai, R.H.; Yamagishi, M.E. Detection of human interchromosomal trans-splicing in sequence databanks. Brief. Bioinform. 2010, 11, 198–209. [Google Scholar] [CrossRef]
- Kim, P.; Yoon, S.; Kim, N.; Lee, S.; Ko, M.; Lee, H.; Kang, H.; Kim, J.; Lee, S. Chimerdb 2.0—A knowledgebase for fusion genes updated. Nucleic Acids Res. 2010, 38, D81–D85. [Google Scholar] [CrossRef]
- McManus, C.J.; Duff, M.O.; Eipper-Mains, J.; Graveley, B.R. Global analysis of trans-splicing in drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 12975–12979. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep rna sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Al-Balool, H.H.; Weber, D.; Liu, Y.; Wade, M.; Guleria, K.; Nam, P.L.; Clayton, J.; Rowe, W.; Coxhead, J.; Irving, J.; et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011, 21, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Wei, Y.; Kang, Y.; Landweber, L.F. Detection of a common chimeric transcript between human chromosomes 7 and 16. Biol. Direct 2012, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Kuo, H.C. The trans-spliced long noncoding rna tsrmst impedes human embryonic stem cell differentiation through wnt5a-mediated inhibition of the epithelial-to-mesenchymal transition. Stem Cells 2016, 34, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Cabili, M.N.; Rinn, J.; Raj, A. Linc-hoxa1 is a noncoding rna that represses hoxa1 transcription in cis. Genes Dev. 2013, 27, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jorgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncrna fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef]
- Deng, C.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.; Bungert, J.; Qiu, Y.; Huang, S. Hoxblinc rna recruits set1/mll complexes to activate hox gene expression patterns and mesoderm lineage development. Cell Rep. 2016, 14, 103–114. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, J.; Hu, G. Genome-wide methods for investigating long noncoding rnas. Biomed. Pharmacother. 2019, 111, 395–401. [Google Scholar] [CrossRef]
- Petri, R.; Jakobsson, J. Identifying mirna targets using ago-ripseq. Methods Mol. Biol. 2018, 1720, 131–140. [Google Scholar] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.C.; Ilicic, T.; Henriksson, J.; Natarajan, K.N.; Tuck, A.C.; Gao, X.; Buhler, M.; Liu, P.; et al. Single cell rna-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Lukowski, S.W.; Chiu, H.S.; Senabouth, A.; Bruxner, T.J.C.; Christ, A.N.; Palpant, N.J.; Powell, J.E. Single-cell rna-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018, 28, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Tiklova, K.; Bjorklund, A.K.; Lahti, L.; Fiorenzano, A.; Nolbrant, S.; Gillberg, L.; Volakakis, N.; Yokota, C.; Hilscher, M.M.; Hauling, T.; et al. Single-cell rna sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun. 2019, 10, 581. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding rnas in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNAs | PRC2 Protein Partners | Regulatory Function | Refs. |
|---|---|---|---|
| Xist | EZH2, JARID 2 | X-chromosome inactivation | [82] |
| Kcnq1ot1 | EZH2, SUZ12 | Kcnq1 inactivation | [92] |
| Meg3 | EZH2, JARID2 | ESC subset of genes inactivation | [93,94] |
| tsRMST | SUZ12 | Pluripotency-associated genes activation (NANOG, POU5F1, SOX2) and cell commitment genes inhibition (GATA6, PAX6) | [95] |
| Linc-HOXA1 | SUZ12 | Hoxa inactivation | [96] |
| T-UCstem1 | SUZ12 | Global de-repression of bivalent chromatin-associated genes | [13] |
| Bvht | SUZ12 | Mesp1 activation | [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorenzano, A.; Pascale, E.; Patriarca, E.J.; Minchiotti, G.; Fico, A. LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells. Epigenomes 2019, 3, 14. https://doi.org/10.3390/epigenomes3030014
Fiorenzano A, Pascale E, Patriarca EJ, Minchiotti G, Fico A. LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells. Epigenomes. 2019; 3(3):14. https://doi.org/10.3390/epigenomes3030014
Chicago/Turabian StyleFiorenzano, Alessandro, Emilia Pascale, Eduardo Jorge Patriarca, Gabriella Minchiotti, and Annalisa Fico. 2019. "LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells" Epigenomes 3, no. 3: 14. https://doi.org/10.3390/epigenomes3030014
APA StyleFiorenzano, A., Pascale, E., Patriarca, E. J., Minchiotti, G., & Fico, A. (2019). LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells. Epigenomes, 3(3), 14. https://doi.org/10.3390/epigenomes3030014