Global DNA Methylation in the Limbic System of Cattle



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
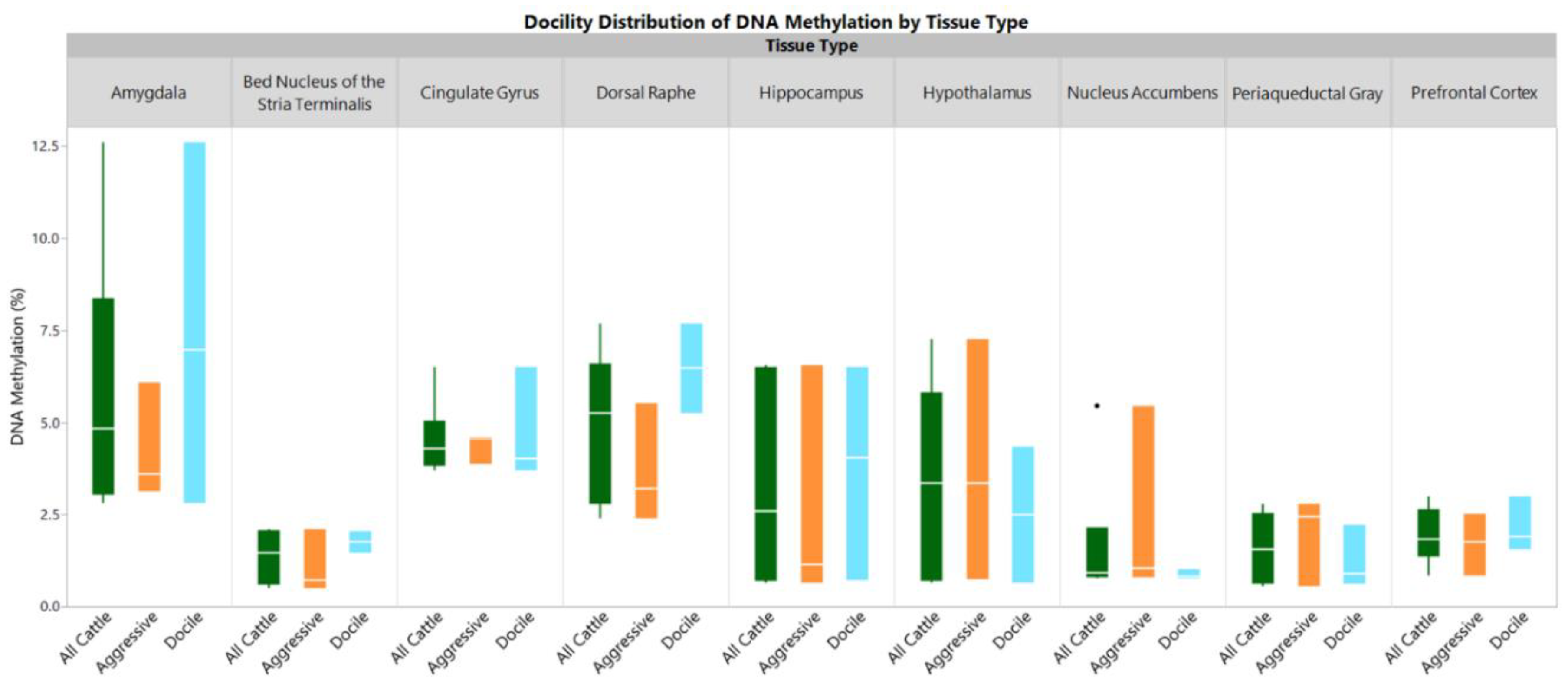
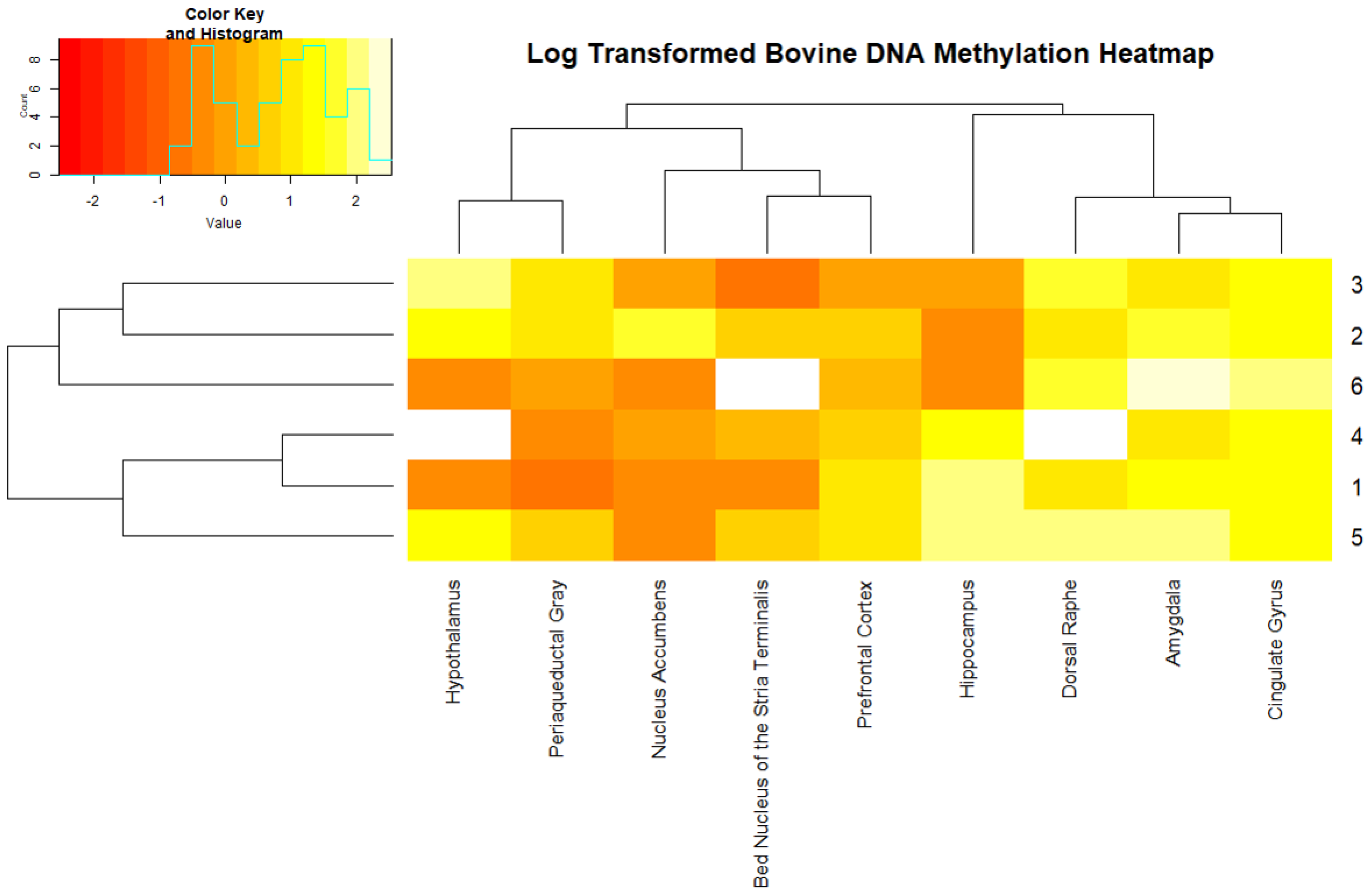
2. Results
3. Discussion
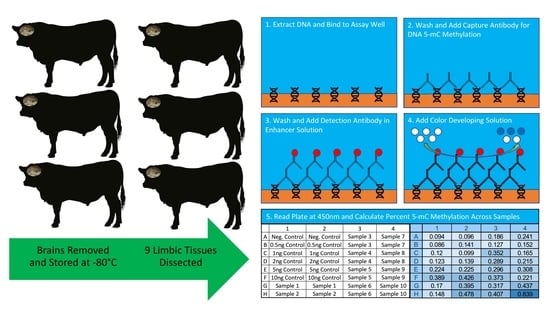
4. Materials and Methods
4.1. Animals and Tissue Collection
4.2. Sample Processing
4.3. Global DNA Methylation Measurement
4.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Murdoch, B.M.; Murdoch, G.K.; Greenwood, S.; McKay, S. Nutritional Influence on Epigenetic Marks and Effect on Livestock Production. Front. Genet. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Meaney, M.J.; Szyf, M. Environmental programming of stress responses through DNA methylation: Life at the interface between a dynamic environment and a fixed genome. Dialogues Clin. Neurosci. 2005, 7, 103–123. [Google Scholar] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.M.; Coleman, P.D.; Rutten, B.P.F.; van den Hove, D.L.A. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Jadavji, N.M.; Deng, L.; Leclerc, D.; Malysheva, O.; Bedell, B.J.; Caudill, M.A.; Rozen, R. Severe methylenetetrahydrofolate reductase deficiency in mice results in behavioral anomalies with morphological and biochemical changes in hippocampus. Mol. Genet. Metab. 2012, 106, 149–159. [Google Scholar] [CrossRef]
- LaPlant, Q.; Vialou, V.; Covington, H.E.; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iniguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Mychasiuk, R.; Harker, A.; Ilnytskyy, S.; Gibb, R. Paternal stress prior to conception alters DNA methylation and behaviour of developing rat offspring. Neuroscience 2013, 241, 100–105. [Google Scholar] [CrossRef]
- Mychasiuk, R.; Ilnytskyy, S.; Kovalchuk, O.; Kolb, B.; Gibb, R. Intensity matters: Brain, behaviour and the epigenome of prenatally stressed rats. Neuroscience 2011, 180, 105–110. [Google Scholar] [CrossRef]
- Mychasiuk, R.; Muhammad, A.; Ilnytskyy, S.; Kolb, B. Persistent gene expression changes in NAc, mPFC, and OFC associated with previous nicotine or amphetamine exposure. Behav. Brain Res. 2013, 256, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.M., Jr.; Toffoli, L.V.; Manfredo, M.H.; Francis-Oliveira, J.; Silva, A.S.; Raquel, H.A.; Martins-Pinge, M.C.; Moreira, E.G.; Fernandes, K.B.; Pelosi, G.G.; et al. Acute stress affects the global DNA methylation profile in rat brain: Modulation by physical exercise. Behav. Brain Res. 2015, 279, 123–128. [Google Scholar] [CrossRef]
- Sable, P.; Randhir, K.; Kale, A.; Chavan-Gautam, P.; Joshi, S. Maternal micronutrients and brain global methylation patterns in the offspring. Nutr. Neurosci. 2015, 18, 30–36. [Google Scholar] [CrossRef]
- Simmons, R.K.; Stringfellow, S.A.; Glover, M.E.; Wagle, A.A.; Clinton, S.M. DNA methylation markers in the postnatal developing rat brain. Brain Res. 2013, 1533, 26–36. [Google Scholar] [CrossRef]
- Toffoli, L.V.; Rodrigues, G.M., Jr.; Oliveira, J.F.; Silva, A.S.; Moreira, E.G.; Pelosi, G.G.; Gomes, M.V. Maternal exposure to fluoxetine during gestation and lactation affects the DNA methylation programming of rat’s offspring: Modulation by folic acid supplementation. Behav. Brain Res. 2014, 265, 142–147. [Google Scholar] [CrossRef]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investig. 2013, 123, 3552–3563. [Google Scholar] [CrossRef]
- Gibbs, J.R.; van der Brug, M.P.; Hernandez, D.G.; Traynor, B.J.; Nalls, M.A.; Lai, S.-L.; Arepalli, S.; Dillman, A.; Rafferty, I.P.; Troncoso, J.; et al. Abundant Quantitative Trait Loci Exist for DNA Methylation and Gene Expression in Human Brain. PLoS Genet. 2010, 6, e1000952. [Google Scholar] [CrossRef]
- Keltner, D.; Kogan, A.; Piff, P.K.; Saturn, S.R. The Sociocultural Appraisals, Values, and Emotions (SAVE) Framework of Prosociality: Core Processes from Gene to Meme. Annu. Rev. Psychol. 2014, 65, 425–460. [Google Scholar] [CrossRef]
- Morgane, P.J.; Galler, J.R.; Mokler, D.J. A review of systems and networks of the limbic forebrain/limbic midbrain. Prog. Neurobiol. 2005, 75, 143–160. [Google Scholar] [CrossRef]
- Mega, F.; de Meireles, A.L.F.; Piazza, F.V.; Spindler, C.; Segabinazi, E.; dos Santos Salvalaggio, G.; Achaval, M.; Marcuzzo, S. Paternal physical exercise demethylates the hippocampal DNA of male pups without modifying the cognitive and physical development. Behav. Brain Res. 2018, 348, 1–8. [Google Scholar] [CrossRef]
- Spindler, C.; Segabinazi, E.; Meireles, A.; Piazza, F.; Mega, F.; dos Santos Salvalaggio, G.; Achaval, M.; Elsner, V.; Marcuzzo, S. Paternal physical exercise modulates global DNA methylation status in the hippocampus of male rat offspring. Neural. Regen. Res. 2019, 14, 491–500. [Google Scholar] [CrossRef]
- Chastain, L.G.; Franklin, T.; Gangisetty, O.; Cabrera, M.A.; Mukherjee, S.; Shrivastava, P.; Jabbar, S.; Sarkar, D.K. Early life alcohol exposure primes hypothalamic microglia to later-life hypersensitivity to immune stress: Possible epigenetic mechanism. Neuropsychopharmacology 2019. [Google Scholar] [CrossRef]
- Mendonça, A.S.; Braga, T.F.; Melo, E.O.; Dode, M.A.; Franco, M.M. Distribution of 5-methylcytosine and 5-hydroxymethylcytosine in bovine fetal tissue of the placenta. Pesqui. Veterinária Bras. 2018, 38, 2012–2018. [Google Scholar]
- Tools of the trade. Nature 2008, 454, 796. [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Kremer, D.; Metzger, S.; Kolb-Bachofen, V.; Kremer, D. Quantitative measurement of genome-wide DNA methylation by a reliable and cost-efficient enzyme-linked immunosorbent assay technique. Anal. Biochem. 2012, 422, 74–78. [Google Scholar] [CrossRef]
- Chowdhury, B.; Cho, I.-H.; Irudayaraj, J. Technical advances in global DNA methylation analysis in human cancers. J. Biol. Eng. 2017, 11, 10. [Google Scholar] [CrossRef]
- Gan, S.D.; Patel, K.R. Enzyme immunoassay and enzyme-linked immunosorbent assay. J. Invest. Derm. 2013, 133, e12. [Google Scholar] [CrossRef]
- Warnes, G.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. gplots: Various R Programming Tools for Plotting Data. R package version 3.0.1. The Comprehensive R Archive Network. 2016. Available online: http://cran.r-project.org/web/packages/gplots/ (accessed on 17 October 2018).
- Chi, J.T.; Chi, E.C.; Baraniuk, R.G. K-POD: A method for k-means clustering of missing data. Am. Stat. 2016, 70, 91–99. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantrell, B.; Lachance, H.; Murdoch, B.; Sjoquist, J.; Funston, R.; Weaber, R.; McKay, S. Global DNA Methylation in the Limbic System of Cattle. Epigenomes 2019, 3, 8. https://doi.org/10.3390/epigenomes3020008
Cantrell B, Lachance H, Murdoch B, Sjoquist J, Funston R, Weaber R, McKay S. Global DNA Methylation in the Limbic System of Cattle. Epigenomes. 2019; 3(2):8. https://doi.org/10.3390/epigenomes3020008
Chicago/Turabian StyleCantrell, Bonnie, Hannah Lachance, Brenda Murdoch, Julia Sjoquist, Richard Funston, Robert Weaber, and Stephanie McKay. 2019. "Global DNA Methylation in the Limbic System of Cattle" Epigenomes 3, no. 2: 8. https://doi.org/10.3390/epigenomes3020008
APA StyleCantrell, B., Lachance, H., Murdoch, B., Sjoquist, J., Funston, R., Weaber, R., & McKay, S. (2019). Global DNA Methylation in the Limbic System of Cattle. Epigenomes, 3(2), 8. https://doi.org/10.3390/epigenomes3020008