An Orphan CpG Island Drives Expression of a let-7 miRNA Precursor with an Important Role in Mouse Development

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
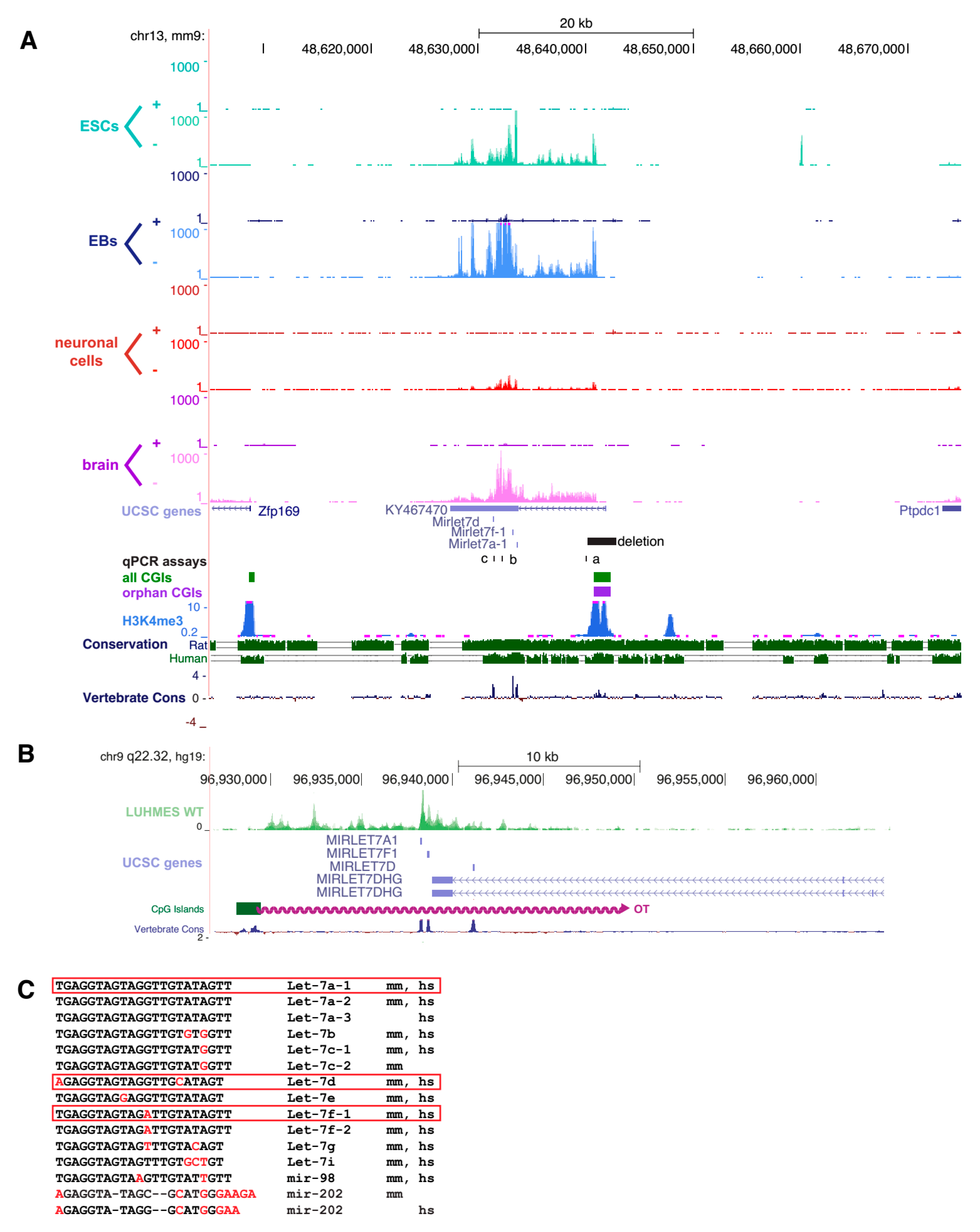
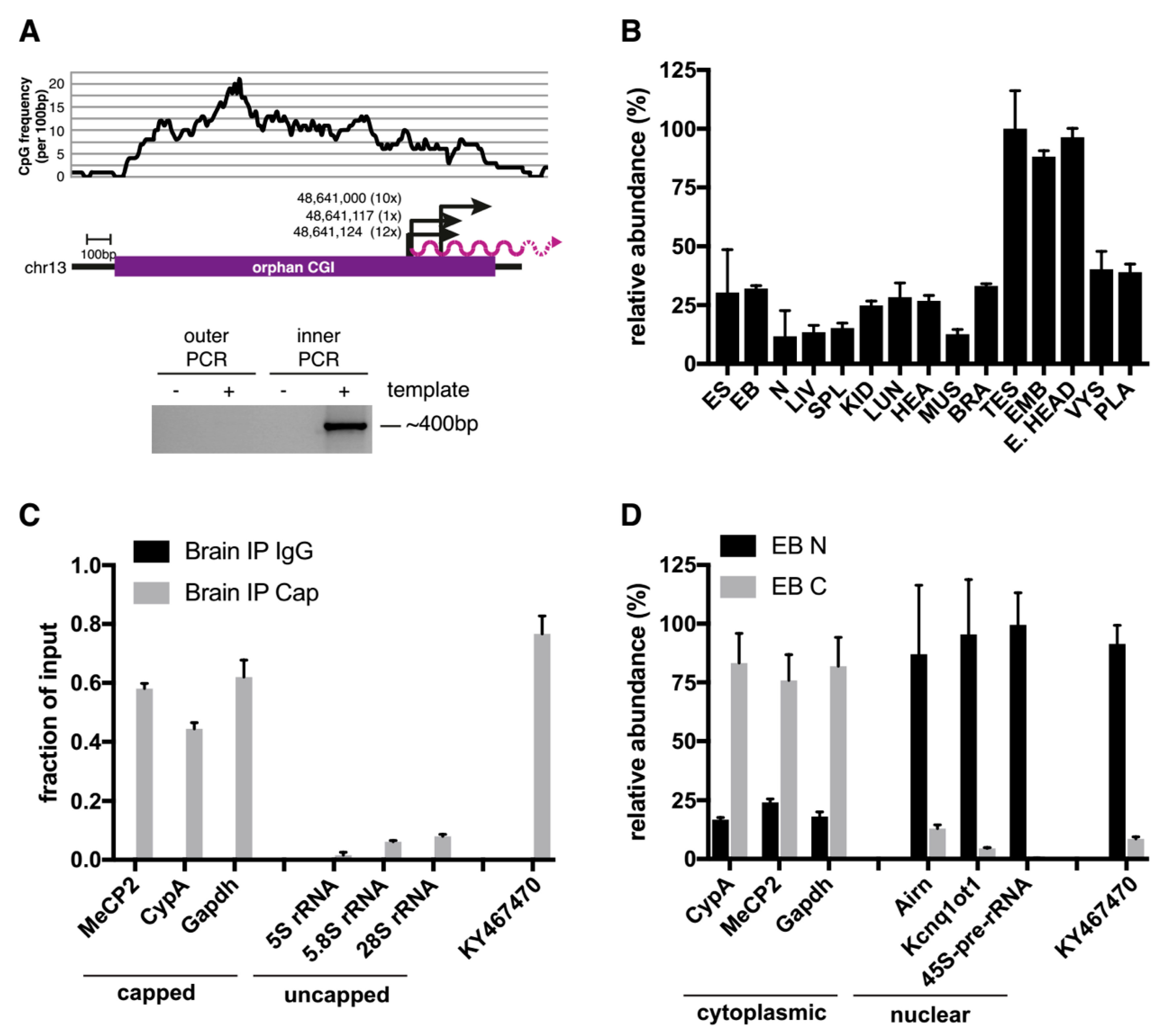
2.1. An Orphan CpG Island Serves as a Promoter for a Conserved miRNA Precursor
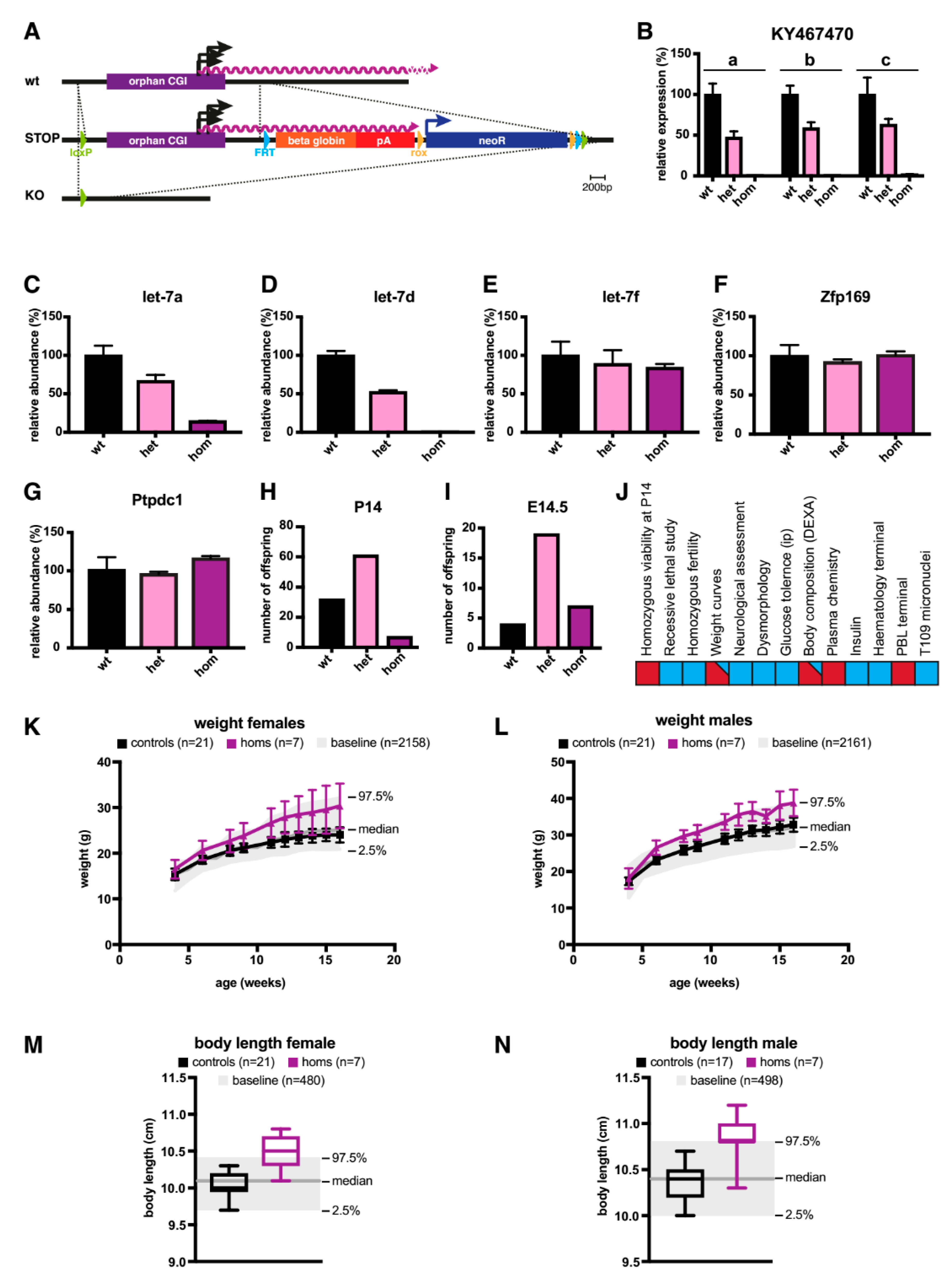
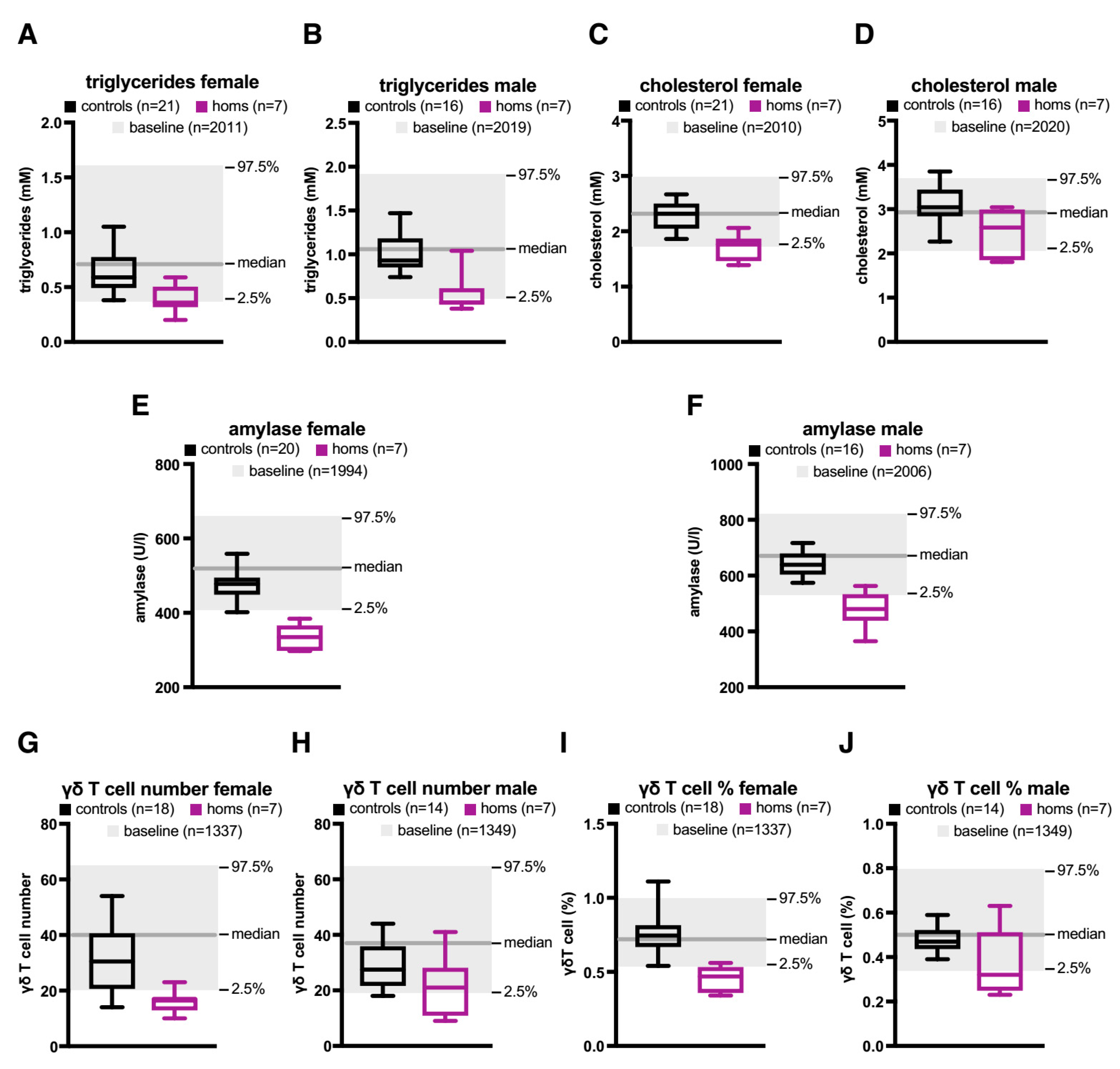
2.2. A Knock-Out (KO) of the Orphan CGI in Mice Abrogates Expression of let-7 Subtypes
3. Discussion
4. Materials and Methods
4.1. ESC Differentiation
4.2. ESC Targeting and Generation of KO Mice
4.3. Mice
4.4. Phenotypic Analysis of Mice
4.5. RNA Extraction
4.6. qPCR Analysis of cDNA
4.7. 5′ RACE
4.8. RNA Seq
4.9. Cap-IP
4.10. Nuclear/Cytoplasmic Fractionation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Zwart, R.; Sleutels, F.; Wutz, A.; Schinkel, A.H.; Barlow, D.P. Bidirectional action of the Igf2r imprint control element on upstream and downstream imprinted genes. Genes Dev. 2001, 15, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Sleutels, F.; Tjon, G.; Ludwig, T.; Barlow, D.P. Imprinted silencing of Slc22a2 and Slc22a3 does not need transcriptional overlap between Igf2r and Air. EMBO J. 2003, 22, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V.; Şenergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012, 338, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.M.; Newman, M.; Parker, J.S.; Morin-Kensicki, E.M.; Wright, T.; Hammond, S.M. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006, 20, 2202–2207. [Google Scholar] [CrossRef]
- Wulczyn, F.G.; Smirnova, L.; Rybak, A.; Brandt, C.; Kwidzinski, E.; Ninnemann, O.; Strehle, M.; Seiler, A.; Schumacher, S.; Nitsch, R. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007, 21, 415–426. [Google Scholar] [CrossRef]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Lancman, J.J.; Caruccio, N.C.; Harfe, B.D.; Pasquinelli, A.E.; Schageman, J.J.; Pertsemlidis, A.; Fallon, J.F. Analysis of the regulation of lin-41 during chick and mouse limb development. Dev. Dyn. 2005, 234, 948–960. [Google Scholar] [CrossRef]
- Schulman, B.R.; Esquela-Kerscher, A.; Slack, F.J. Reciprocal expression of lin-41 and the microRNAs let-7 and mir-125 during mouse embryogenesis. Dev. Dyn. 2005, 234, 1046–1054. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef]
- White, J.K.; Gerdin, A.K.; Karp, N.A.; Ryder, E.; Buljan, M.; Bussell, J.N.; Salisbury, J.; Clare, S.; Ingham, N.J.; Podrini, C.; et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 2013, 154, 452–464. [Google Scholar] [CrossRef]
- Wutz, A.; Smrzka, O.W.; Schweifer, N.; Schellander, K.; Wagner, E.F.; Barlow, D.P. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 1997, 389, 745–749. [Google Scholar] [CrossRef]
- Lyle, R.; Watanabe, D.; Te Vruchte, D.; Lerchner, W.; Smrzka, O.W.; Wutz, A.; Schageman, J.; Hahner, L.; Davies, C.; Barlow, D.P. The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat. Genet. 2000, 25, 19–21. [Google Scholar] [CrossRef]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Knight, S.W.; Bass, B.L. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; McLachlan, J.; Pasquinelli, A.E.; Balint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef]
- Worringer, K.A.; Rand, T.A.; Hayashi, Y.; Sami, S.; Takahashi, K.; Tanabe, K.; Narita, M.; Srivastava, D.; Yamanaka, S. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 2014, 14, 40–52. [Google Scholar] [CrossRef]
- Zhu, H.; Shah, S.; Shyh-Chang, N.; Shinoda, G.; Einhorn, W.S.; Viswanathan, S.R.; Takeuchi, A.; Grasemann, C.; Rinn, J.L.; Lopez, M.F.; et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat. Genet. 2010, 42, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Slack, F.J. MicroRNA control of lifespan and metabolism. Cell Cycle 2006, 5, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Jackson, A.U.; Gieger, C.; Schumacher, F.R.; Berndt, S.I.; Sanna, S.; Eyheramendy, S.; Voight, B.F.; Butler, J.L.; Guiducci, C.; et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008, 40, 584–591. [Google Scholar] [CrossRef]
- Bain, G.; Kitchens, D.; Yao, M.; Huettner, J.E.; Gottlieb, D.I. Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 1995, 168, 342–357. [Google Scholar] [CrossRef]
- Li, M.; Pevny, L.; Lovell-Badge, R.; Smith, A. Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr. Biol. 1998, 8, 971–974. [Google Scholar] [CrossRef]
- Kurbatova, N.; Mason, J.C.; Morgan, H.; Meehan, T.F.; Karp, N.A. PhenStat: A Tool Kit for Standardized Analysis of High Throughput Phenotypic Data. PLoS ONE 2015, 10, e0131274. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Scholz, D.; Pöltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Cholewa-Waclaw, J.; Davies, F.C.; Paton, K.M.; Chaligne, R.; Heard, E.; Abbott, C.M.; Bird, A.P. Efficient and versatile CRISPR engineering of human neurons in culture to model neurological disorders. Wellcome Open Res. 2016, 1, 13. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Seidl, C.I.; Stricker, S.H.; Barlow, D.P. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. Embo J. 2006, 25, 3565–3575. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koerner, M.V.; Chhatbar, K.; Webb, S.; Cholewa-Waclaw, J.; Selfridge, J.; De Sousa, D.; Skarnes, B.; Rosen, B.; Thomas, M.; Bottomley, J.; et al. An Orphan CpG Island Drives Expression of a let-7 miRNA Precursor with an Important Role in Mouse Development. Epigenomes 2019, 3, 7. https://doi.org/10.3390/epigenomes3010007
Koerner MV, Chhatbar K, Webb S, Cholewa-Waclaw J, Selfridge J, De Sousa D, Skarnes B, Rosen B, Thomas M, Bottomley J, et al. An Orphan CpG Island Drives Expression of a let-7 miRNA Precursor with an Important Role in Mouse Development. Epigenomes. 2019; 3(1):7. https://doi.org/10.3390/epigenomes3010007
Chicago/Turabian StyleKoerner, Martha V., Kashyap Chhatbar, Shaun Webb, Justyna Cholewa-Waclaw, Jim Selfridge, Dina De Sousa, Bill Skarnes, Barry Rosen, Mark Thomas, Joanna Bottomley, and et al. 2019. "An Orphan CpG Island Drives Expression of a let-7 miRNA Precursor with an Important Role in Mouse Development" Epigenomes 3, no. 1: 7. https://doi.org/10.3390/epigenomes3010007
APA StyleKoerner, M. V., Chhatbar, K., Webb, S., Cholewa-Waclaw, J., Selfridge, J., De Sousa, D., Skarnes, B., Rosen, B., Thomas, M., Bottomley, J., Ramirez-Solis, R., Lelliott, C., Adams, D. J., & Bird, A. (2019). An Orphan CpG Island Drives Expression of a let-7 miRNA Precursor with an Important Role in Mouse Development. Epigenomes, 3(1), 7. https://doi.org/10.3390/epigenomes3010007