Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bromodomain and Extraterminal Inhibitors in Pancreatic Cancer
2.1. Role and Effects of Bromodomain and Extraterminal Inhibitors in Pancreatic Cancer
2.2. Bromodomain and Extraterminal Inhibition and Metastasis in Pancreatic Cancer
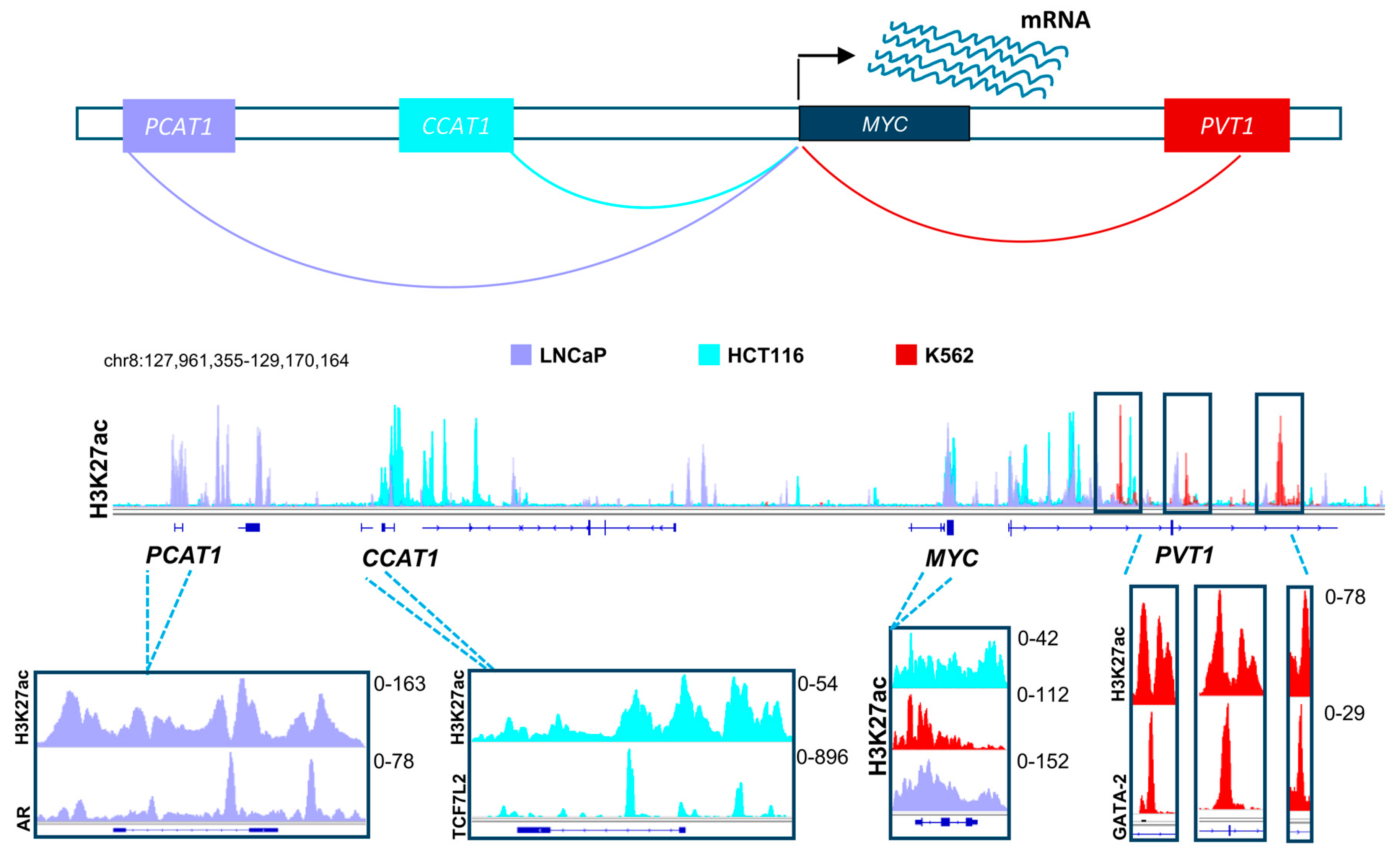
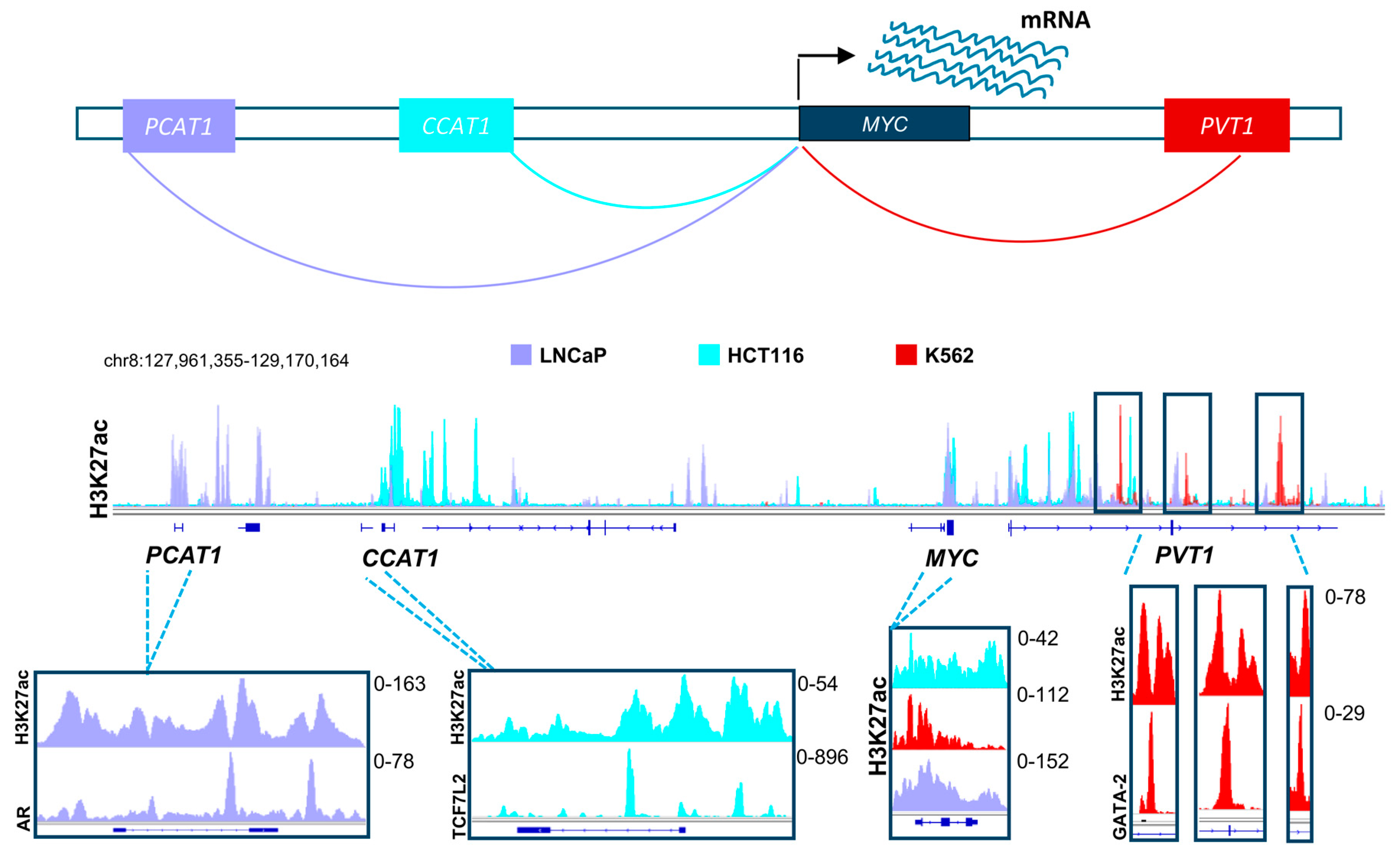
2.3. Effect of Bromodomain and Extraterminal Inhibition on Distal Regulatory Elements
3. Histone Deacetylase Inhibitors in Pancreatic Cancer
3.1. Role and Effects of Histone Deacetylase Inhibitors in Pancreatic Cancer
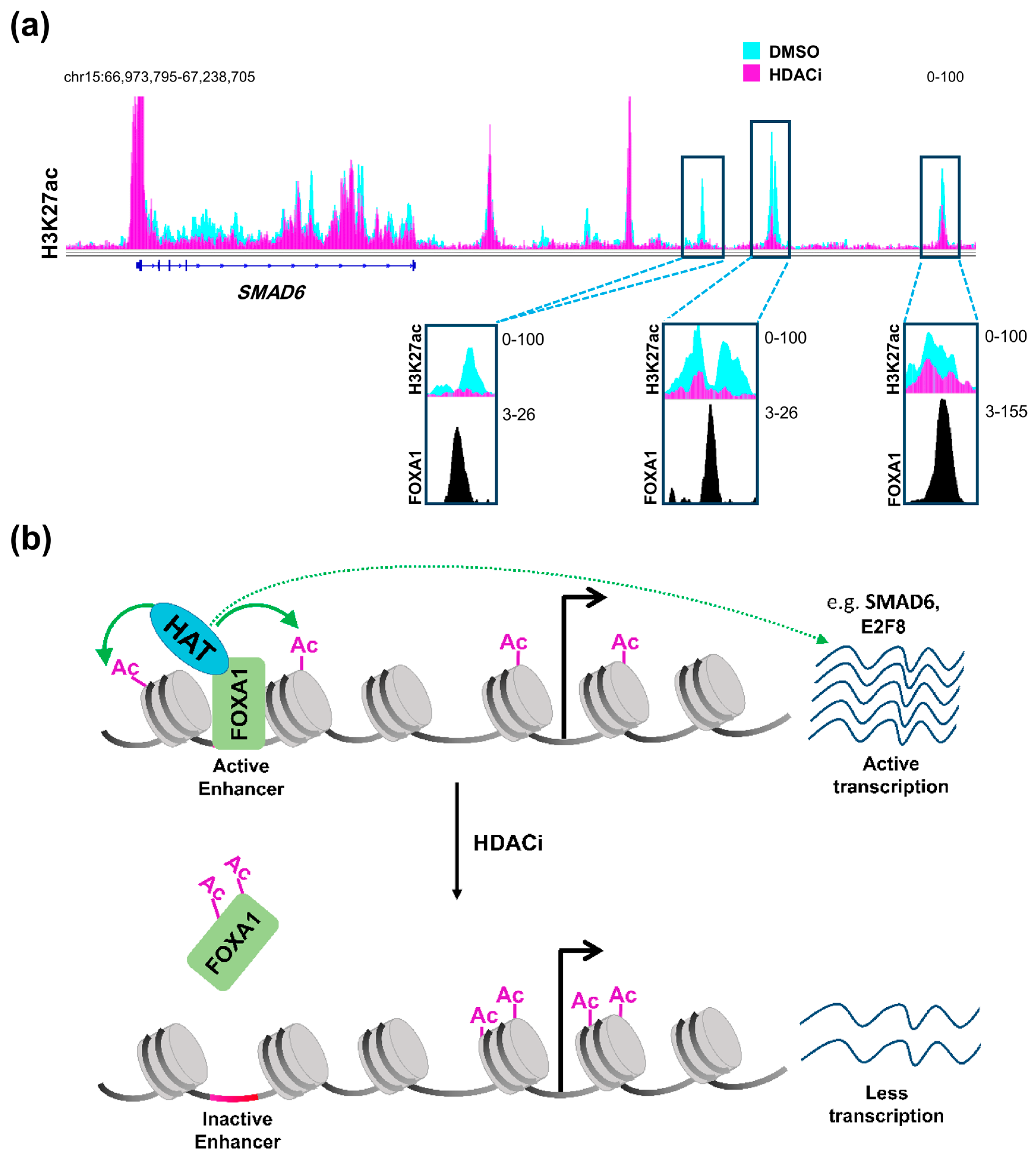
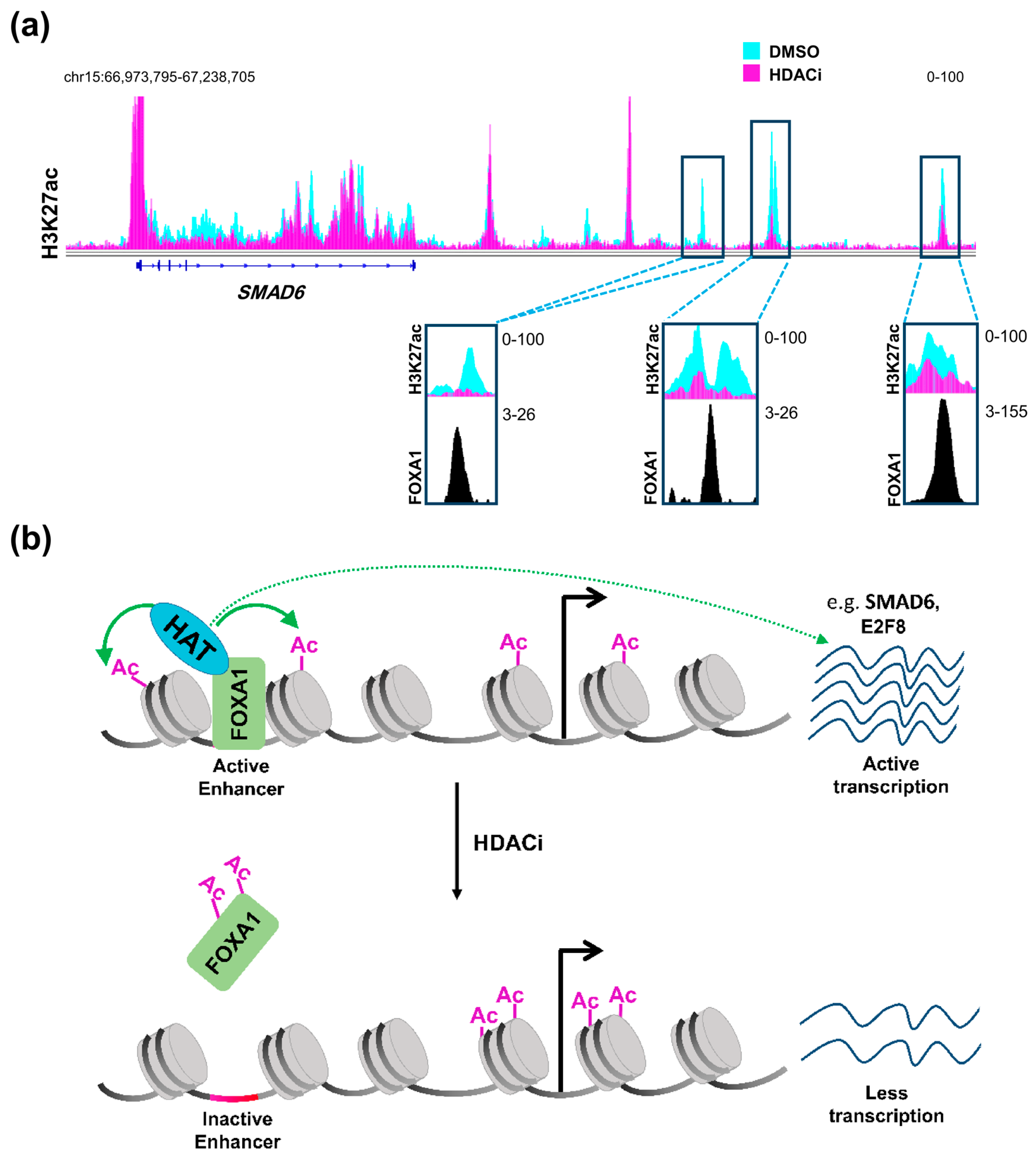
3.2. Histone Deacetylase Inhibition Role at Distal Regulatory Elements
4. Enhancers as an Optimal Paradigm for Therapeutic Targeting of Pancreatic Cancer
5. Targeting Transcription Factors in Pancreatic Cancer: A Code for Specificity
6. Conclusions: Unconventional Epigenetic Agents Should Be Used in Unconventional Ways
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A. Parameters for Data Analysis Used in Figure Generation
References
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann. Oncol. 2017, 28, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Jakel, C.; Bergmann, F.; Toth, R.; Assenov, Y.; van der Duin, D.; Strobel, O.; Hank, T.; Kloppel, G.; Dorrell, C.; Grompe, M.; et al. Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat. Commun. 2017, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Parikh, H.; Xiao, W.; Hoskins, J.W.; Pflicke, H.; Liu, X.; Collins, I.; Zhou, W.; Wang, Z.; Powell, J.; et al. An integrated transcriptome and epigenome analysis identifies a novel candidate gene for pancreatic cancer. BMC Med. Genom. 2013, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- McCleary-Wheeler, A.L.; Lomberk, G.A.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Iguchi, E.; Safgren, S.L.; Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Pancreatic cancer, a mis-interpreter of the epigenetic language. Yale J. Biol. Med. 2016, 89, 575–590. [Google Scholar] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Gene Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Gene Dev. 2015, 29, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of valproic acid in combination with S-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014, 34, 5187–5191. [Google Scholar] [PubMed]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, E.; Kerner, Z.; Nanda, N.; Ahuja, N. Epigenetic therapy in gastrointestinal cancer: The right combination. Ther. Adv. Gastroenterol. 2016, 9, 560–579. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.A.; Iovanna, J.; Urrutia, R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics 2016, 8, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef] [PubMed]
- Silverman, B.R.; Shi, J. Alterations of epigenetic regulators in pancreatic cancer and their clinical implications. Int. J. Mol. Sci. 2016, 17, 2138. [Google Scholar] [CrossRef] [PubMed]
- Quilichini, E.; Haumaitre, C. Implication of epigenetics in pancreas development and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Klieser, E.; Swierczynski, S.; Mayr, C.; Schmidt, J.; Neureiter, D.; Kiesslich, T.; Illig, R. Role of histone deacetylases in pancreas: Implications for pathogenesis and therapy. World J. Gastrointest. Oncol. 2015, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpis, N.; Karatzas, T.; Nikolidakis, L.; Kostakis, I.D.; Garmpi, A.; Karamaroudis, S.; Boutsikos, G.; Damaskou, Z.; Kostakis, A.; et al. Histone deacetylase (HDAC) inhibitors: Current evidence for therapeutic activities in pancreatic cancer. Anticancer Res. 2015, 35, 3129–3135. [Google Scholar] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [PubMed]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed]
- French, C.A. Pathogenesis of NUT midline carcinoma. Ann. Rev. Pathol. 2012, 7, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Al Diffalha, S.; Al Aukla, N.; Hasan, S.; Dickinson, S.; Khalil, F. NUT midline carcinoma: A rare malignancy. Cancer Control 2017, 24, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.G.; French, C.A.; Cameron, M.J.; Fletcher, C.D.; Jackman, D.M.; Lathan, C.S.; Sholl, L.M. Pathologic characteristics of NUT midline carcinoma arising in the mediastinum. Am. J. Surg. Pathol. 2012, 36, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Tian, M.; Li, X.; Li, J.J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI underlies a role for BET bromodomains in pancreatic cancer growth and the tumor microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Bigonnet, M.; Gayet, O.; Loncle, C.; Maignan, A.; Gilabert, M.; Moutardier, V.; Garcia, S.; Turrini, O.; Delpero, J.R.; et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: Implications for individualized medicine efforts. EMBO Mol. Med. 2017, 9, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Uccello, M.; Moschetta, M.; Mak, G.; Alam, T.; Henriquez, C.M.; Arkenau, H.T. Towards an optimal treatment algorithm for metastatic pancreatic ductal adenocarcinoma (PDA). Curr. Oncol. 2018, 25, e90–e94. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.J.; Bayles, I.; Funnell, A.P.W.; Miller, T.E.; Saiakhova, A.; Lizardo, M.M.; Bartels, C.F.; Kapteijn, M.Y.; Hung, S.; Mendoza, A.; et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat. Med. 2018, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; De Leo, A.; Wang, Z.; Kerekovic, A.; Hills, R.; Lieberman, P.M. BET-inhibitors disrupt Rad21-dependent conformational control of KSHV latency. PLoS Pathog. 2017, 13, e1006100. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Zhang, F.; Jaganathan, A.; Sharma, R.; Zhang, Q.; Konuma, T.; Shen, T.; Lee, J.Y.; Ren, C.; Chen, C.H.; et al. Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol. Cell 2017, 65, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Gilgenast, T.G.; Bartman, C.R.; Edwards, C.R.; Stonestrom, A.J.; Huang, P.; Emerson, D.J.; Evans, P.; Werner, M.T.; Keller, C.A.; et al. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 2017, 66, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Oh, S.; Yoo, K.H. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol. Cells 2017, 40, 169–177. [Google Scholar] [PubMed]
- Kaiser, V.B.; Semple, C.A. When TADs go bad: Chromatin structure and nuclear organisation in human disease. F1000Research 2017, 6, 314. [Google Scholar] [CrossRef] [PubMed]
- Najafova, Z.; Tirado-Magallanes, R.; Subramaniam, M.; Hossan, T.; Schmidt, G.; Nagarajan, S.; Baumgart, S.J.; Mishra, V.K.; Bedi, U.; Hesse, E.; et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017, 45, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Anyansi, C.; Hu, X.; Xu, L.; Xiong, L.; Tang, W.; Mok, M.T.S.; Cheng, C.; Fan, X.; Gerstein, M.; et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017, 49, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Nagarajan, S.; Baumgart, S.J.; Kosinsky, R.L.; Najafova, Z.; Kari, V.; Hennion, M.; Indenbirken, D.; Bonn, S.; Grundhoff, A.; et al. RNF40 regulates gene expression in an epigenetic context-dependent manner. Genome Biol. 2017, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Fontanals-Cirera, B.; Hasson, D.; Vardabasso, C.; Di Micco, R.; Agrawal, P.; Chowdhury, A.; Gantz, M.; de Pablos-Aragoneses, A.; Morgenstern, A.; Wu, P.; et al. Harnessing BET inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene. Mol. Cell 2017, 68, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Kannan, A.; Kern, M.; Moreno, M.A.; Vural, E.; Stack, B., Jr.; Suen, J.Y.; Tackett, A.J.; Gao, L. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 2015, 10, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.H.; Johnsen, S.A. Super enhancers—New analyses and perspectives on the low hanging fruit. Transcription 2018, 9, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Gelato, K.A.; Schockel, L.; Klingbeil, O.; Ruckert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Heroult, M.; Lejeune, P.; Monning, U.; et al. Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Muhar, M.; Ebert, A.; Neumann, T.; Umkehrer, C.; Jude, J.; Wieshofer, C.; Rescheneder, P.; Lipp, J.J.; Herzog, V.A.; Reichholf, B.; et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 2018, 360, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harbor Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Vancurova, I.; Uddin, M.M.; Zou, Y.; Vancura, A. Combination therapies targeting HDAC and IKK in solid tumors. Trends Pharmacol. Sci. 2018, 39, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wang, W.H.; Wu, W.Y.; Hsu, C.C.; Wei, L.R.; Wang, S.F.; Hsu, Y.W.; Liaw, C.C.; Tsai, W.C. Novel histone deacetylase inhibitor AR-42 exhibits antitumor activity in pancreatic cancer cells by affecting multiple biochemical pathways. PLoS ONE 2017, 12, e0183368. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Qiao, Z.; Wang, Y.; Kuai, Q.; Li, C.; Wang, Y.; Jiang, X.; Wang, X.; Li, W.; He, M.; et al. Chidamide inhibits aerobic metabolism to induce pancreatic cancer cell growth arrest by promoting Mcl-1 degradation. PLoS ONE 2016, 11, e0166896. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Manfredi, M.; Brandi, J.; Buzzi, A.; Conte, E.; Pacchiana, R.; Cecconi, D.; Marengo, E.; Donadelli, M. Trichostatin A alters cytoskeleton and energy metabolism of pancreatic adenocarcinoma cells: An in depth proteomic study. J. Cell. Biochem. 2018, 119, 2696–2707. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, J.; Zhao, J.; Yun, W.; Xie, C.; Taub, J.W.; Azmi, A.; Mohammad, R.M.; Dong, Y.; Kong, W.; et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS ONE 2012, 7, e52095. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by histone deacetylase inhibition confers elimination of pancreatic cancer stem cells in combination with gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [PubMed]
- Samulitis, B.K.; Pond, K.W.; Pond, E.; Cress, A.E.; Patel, H.; Wisner, L.; Patel, C.; Dorr, R.T.; Landowski, T.H. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol. Ther. 2015, 16, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, L.; Kang, S.G.; Lu, Y.; Yang, Z.; Huynh, T.; Chen, C.; Zhou, R.; Guo, M.; Zhao, Y. Gd-metallofullerenol nanomaterial suppresses pancreatic cancer metastasis by inhibiting the interaction of histone deacetylase 1 and metastasis-associated protein 1. ACS Nano 2015, 9, 6826–6836. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.J.; Richmond, P.A.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; Wheeler, G.E.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018, 46, 1756–1776. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Ling, K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys27 acetylation in the BIM gene promoter and enhancers. Oncol. Lett. 2017, 14, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.B.; Tanaka, Y.; Kim, Y.J.; Xie, P.; Zhang, M.Q.; Park, I.H.; Kim, T.H. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 2015, 13, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Zhang, B.; Li, Y.; King, J.L.; Tian, R.; Xia, S.; Schiavon, C.R.; Dong, J.T. HDAC-mediated deacetylation of KLF5 associates with its proteasomal degradation. Biochem. Biophys. Res. Commun. 2018, 500, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Beier, U.H.; Akimova, T.; Dahiya, S.; Han, R.; Samanta, A.; Levine, M.H.; Hancock, W.W. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am. J. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Cirillo, L.A. Stable chromatin binding prevents FoxA acetylation, preserving FoxA chromatin remodeling. J. Biol. Chem. 2010, 285, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Gotze, S.; Coersmeyer, M.; Muller, O.; Sievers, S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int. J. Oncol. 2014, 45, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhou, X.; Xia, Q.; Shen, J.; Yan, J.; Zhu, J.; Li, X.; Shu, M. Long non-coding RNA CCAT1 that can be activated by c-Myc promotes pancreatic cancer cell proliferation and migration. Am. J. Transl. Res. 2016, 8, 5444–5454. [Google Scholar] [PubMed]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Cyrta, J.; Murphy, D.A.; Walczak, E.G.; Ran, L.; Agrawal, P.; Xie, Y.; Chen, Y.; Wang, S.; Zhan, Y.; et al. Aberrant Activation of a gastrointestinal transcriptional circuit in prostate cancer mediates castration resistance. Cancer Cell 2017, 32, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.L.; Tuzova, A.V.; Bolton, E.M.; Lynch, T.H.; Perry, A.S. Long noncoding RNAs and prostate carcinogenesis: The missing ‘linc’? Trends Mol. Med. 2014, 20, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. Epigenetics: The first 25 centuries. Philos. Trans. R. Soc. B 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Huang, X.; Bellin, R.; Faivre, E.J.; Hessler, P.; Lam, L.; Bui, M.-H.; Wilcox, D.M.; Uziel, T.; Ferguson, D.C.; et al. ABBV-744, a first-in-class and highly selective inhibitor of the second beomodomain of BET family proteins, displays robust activities in preclinical models of acute myelogenous leukemia. In Proceedings of the AACR Annual Meeting, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Peng, X.L.; So, K.K.; He, L.; Zhao, Y.; Zhou, J.; Li, Y.; Yao, M.; Xu, B.; Zhang, S.; Yao, H.; et al. MyoD- and FoxO3-mediated hotspot interaction orchestrates super-enhancer activity during myogenic differentiation. Nucleic Acids Res. 2017, 45, 8785–8805. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.; Hughes, J.R.; Babbs, C.; Davies, J.O.J.; Graham, B.J.; Hanssen, L.; Kassouf, M.T.; Marieke Oudelaar, A.M.; Sharpe, J.A.; Suciu, M.C.; et al. Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 2016, 48, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y.; Willi, M.; HyunYoo, K.; Zeng, X.; Wang, C.; Metser, G.; Hennighausen, L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016, 48, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Zhang, J.; Fu, Q.; Wei, T.; Sun, X.; Chen, Q.; Yang, J.; Bai, X.; Liang, T. Hypoxia-inducible factor-2αpromotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol. Cancer 2017, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Macgregor-Das, A.; Saunders, T.; Whittle, M.C.; Makohon-Moore, A.; Kohutek, Z.A.; Poling, J.; Herbst, B.T.; Javier, B.M.; Cope, L.; et al. Mutant p53 together with TGFβ signaling influence organ-specific hematogenous colonization patterns of pancreatic cancer. Clin. Cancer Res. 2017, 23, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-κ B pathway leads to deregulation of epithelial-mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. J. Tech. Methods Pathol. 2016, 96, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zeng, S.; Zou, Y.; Shi, M.; Qiu, Q.; Xiao, Y.; Chen, G.; Yang, X.; Liang, L.; Xu, H. The suppression of bromodomain and extra-terminal domain inhibits vascular inflammation by blocking NF-κB and MAPK activation. Br. J. Pharmacol. 2017, 174, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Hensel, T.; Giorgi, C.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Buch, T.; Niggli, F.K.; Schafer, B.W.; Burdach, S.; Richter, G.H. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget 2016, 7, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Blum, K.A.; Younes, A.; Abramson, J.S.; Maris, M.B.; Flinn, I.W.; Goy, A.; Border, D.R.; Cooper, M.R.; Sims, R.J. Pharmacodynamic assessment in whole blood for the BET bromodomain inhibitor CPI-0610 of target engagement in patients with progressive lymphoma. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, 5. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdan, F.H.; Johnsen, S.A. Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer. Epigenomes 2018, 2, 8. https://doi.org/10.3390/epigenomes2020008
Hamdan FH, Johnsen SA. Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer. Epigenomes. 2018; 2(2):8. https://doi.org/10.3390/epigenomes2020008
Chicago/Turabian StyleHamdan, Feda H., and Steven A. Johnsen. 2018. "Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer" Epigenomes 2, no. 2: 8. https://doi.org/10.3390/epigenomes2020008