Epigenomics of Plant Responses to Environmental Stress


{kind=link}
Abstract
1. Introduction
2. Epigenetics of DNA Base Modification
3. Epigenetic Regulation of Chromatin Structure
4. Regulation of Gene Expression and Genome Stability
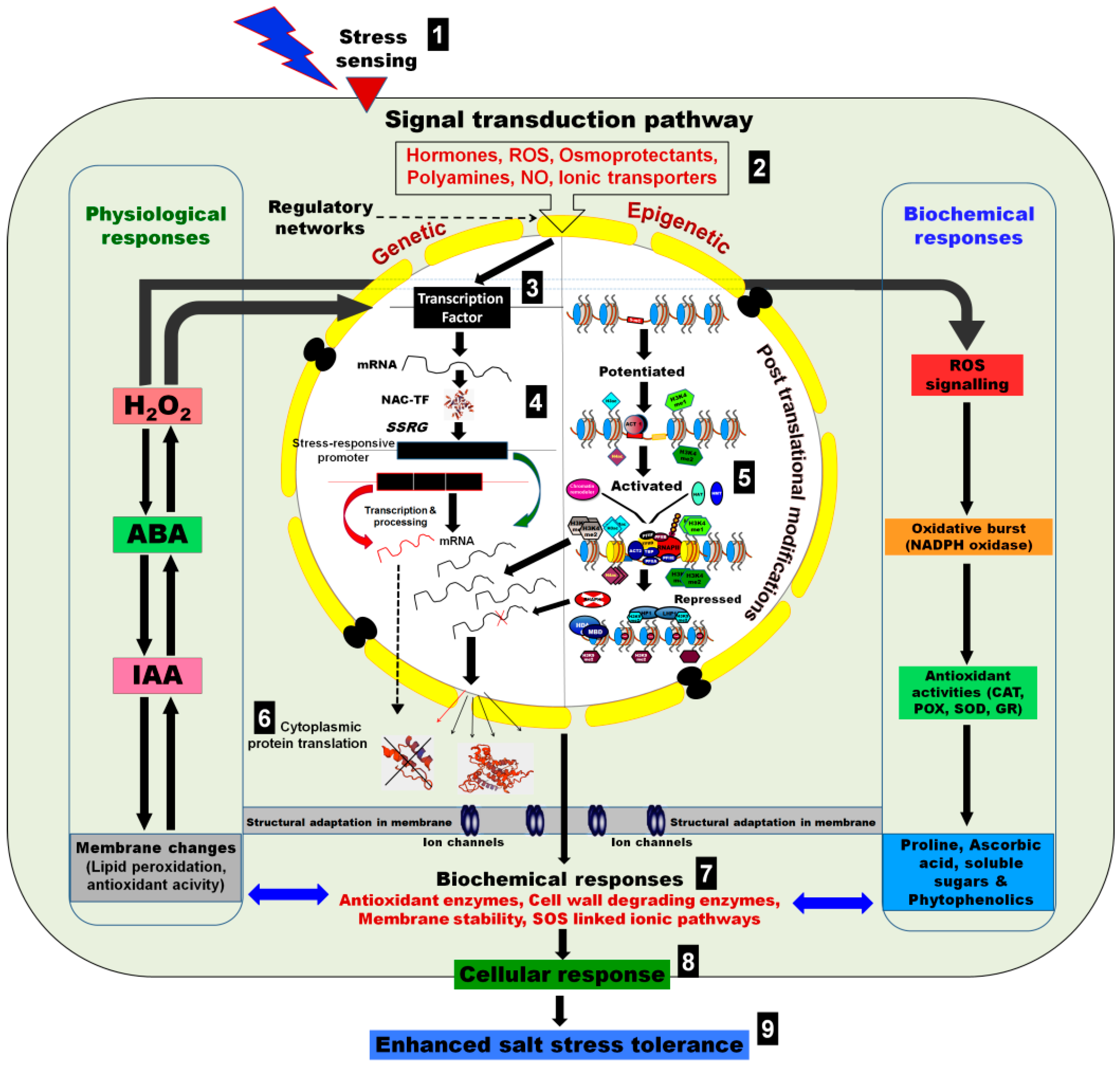
5. Salt-Induced Epigenetic Changes in Crop Plants
6. Transgenerational Inheritance of Epimarks
7. Future Perspectives of Epigenomic Studies
Author Contributions
Conflicts of Interest
References
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.P.; Fabris, D.; Agris, P.F. The RNA modification database, RNAMDB. Nucleic Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F. Bringing order to translation: The contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008, 9, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. tRNA stabilization by modified nucleotides. Biochemistry 2010, 49, 4934–4944. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Pollex, T.; Hanna, K.; Tuorto, F.; Meusburger, M.; Helm, M.; Lyko, F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010, 24, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Laland, K.; Uller, T.; Feldman, M.; Sterelny, K.; Müller, G.B.; Mozcek, A.; Jablonka, E.; Odling-Smee, J.; Wray, G.A.; Hoekstra, H.E.; et al. Does evolutionary theory need a rethink? Nature 2014, 514, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.C.; Zhang, J. The genotype-phenotype map of yeast complex traits: basic parameters and the role of natural selection. Mol. Biol. Evol. 2014, 31, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Epigenetic control of apomixis: A new perspective of an old enigma. Adv. Plants Agric. Res. 2017, 7, 00243. [Google Scholar] [CrossRef]
- Skinner, M.K. Environmental epigenetics and a unified theory of the molecular aspects of evolution: A neo-Lamarckian concept that facilitates neo-Darwinian evolution. Genome Biol. Evol. 2015, 7, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Farias, N.; Ho, N.; Butler, S.; Delaney, L.; Morrison, J.; Shahrzad, S.; Coomber, B.L. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 2015, 26, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Oliver, V.F.; Wang, G.; Zhu, H.; Zack, D.J.; Merbs, S.L.; Qian, J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genom. 2015, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Guo, Z.; Ma, Q.; Xu, Z.; Zhou, Y.; Xu, Z.; Li, Z.; Liu, Y.; Yuan, B.; et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016, 26, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Morales, R.; Agís-Balboa, R.C.; Esteller, M.; Berdasco, M. Epigenetic mechanisms during ageing and neurogenesis as novel therapeutic avenues in human brain disorders. Clin. Epigenet. 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Behera, P. Epigenetics changes in breast cancer: Current aspects in India. J. Bioeng. Biomed. Sci. 2017, 7, 223. [Google Scholar] [CrossRef]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Salt-induced tissue-specific cytosine methylation downregulates expression of HKT genes in contrasting wheat (Triticum aestivum L.) genotypes. DNA Cell Biol. 2017, 36, 283–394. [Google Scholar] [CrossRef] [PubMed]
- Eichten, S.R.; Schmitz, R.J.; Springer, N.M. Epigenetics: Beyond chromatin modifications and complex genetic regulation. Plant Physiol. 2014, 165, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, A. Epigenetic regulation of abiotic stress tolerance in plants. Adv. Plants Agric. Res. 2016, 5, 00179. [Google Scholar] [CrossRef]
- Avramova, Z. Transcriptional “memory” of a stress: Transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J. 2015, 83, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Lauria, M.; Rossi, V. Epigenetic control of gene regulation in plants. Biochim. Biophys. Acta 2011, 1809, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Pikaard, C.S.; MittelstenScheid, O. Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 2014, 6, a019315. [Google Scholar] [CrossRef] [PubMed]
- Gallusci, P.; Hodgman, C.; Teyssier, E.; Seymour, G.B. DNA methylation and chromatin regulation during fleshy fruit development and ripening. Front. Plant Sci. 2016, 7, 807. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Lanciano, S.; Mirouze, M. DNA methylation in rice and relevance for breeding. Epigenomes 2017, 1, 10. [Google Scholar] [CrossRef]
- Li, Y.; Kumar, S.; Qian, W. Active DNA demethylation: Mechanism and role in plant development. Plant Cell Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Schmitz, R.J. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, A.K.; Mohapatra, T. Epigenetics: History, present status and future perspective. Indian J. Genet. Plant Breed. 2017, 77, 445–463. [Google Scholar]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenet. Chromatin 2015, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Q.; Yuan, W.; Cao, Z.; Qi, B.; Kumar, S.; Li, Y.; Qian, W. The cytosolic Fe-S cluster assembly component MET18 is required for the full enzymatic activity of ROS1 in active DNA demethylation. Sci. Rep. 2016, 6, 26443. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzic, M.; Becker, J.D.; Feijo, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.-K. Active DNA demethylation in plants and animals. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Penterman, J.; Zilberman, D.; Huh, J.H.; Ballinger, T.; Henikoff, S.; Fischer, R.L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. USA 2007, 104, 6752–6757. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Steward, N.; Ito, M.; Yamachuchi, Y.; Koizumi, N.; Sano, H. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J. Biol. Chem. 2002, 277, 37741–37746. [Google Scholar] [CrossRef] [PubMed]
- Kou, H.P.; Li, Y.; Song, X.X.; Ou, X.F.; Xing, S.C.; Ma, J.; Von Wettstein, D.; Liu, B. Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J. Plant Physiol. 2011, 168, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Physiological, biochemical, epigenetic and molecular analyses of wheat (Triticum aestivum) genotypes with contrasting salt tolerance. Front. Plant Sci. 2017, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.E.; Mosquna, A.; Ingouff, M.; Sakata, T.; Ohad, N.; Berger, F. Retinoblastoma and its binding partner MSI1 control imprinting in Arabidopsis. PLoS Biol. 2008, 6, e194. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Penn, N.W.; Suwalski, R.; O’Riley, C.; Bojanowski, K.; Yura, R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J. 1972, 126, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Munzel, M.; Muller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, R.M.; Souza, A.L.; Clish, C.B.; Gehring, M. 5-Hydroxymethylcytosine is not present in appreciable quantities in Arabidopsis DNA. Genes Genome Genet. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Casadesus, J.; Low, D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef] [PubMed]
- Ratel, D.; Ravanat, J.L.; Berger, F.; Wion, D. N6-methyladenine: the other methylated base of DNA. BioEssays 2006, 28, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizabal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA Methylation on N-adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Huang, H.; Liu, D.; Cheng, Y.; Liu, X.; Zhang, W.; Yin, R.; Zhang, D.; Zhang, P.; Liu, J.; et al. N6-methyladenine DNA modification in Drosophila. Cell 2015, 161, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Dore, L.C.; et al. N6-Methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Xiong, J.; Yang, Y.; Liu, S.M.; Yuan, B.F.; Feng, Y.Q. Determination of DNA adenine methylation in genomes of mammals and plants by liquid chromatography/mass spectrometry. R. Soc. Chem. Adv. 2015, 5, 64046–64054. [Google Scholar] [CrossRef]
- Bartholomew, B. Regulating the chromatin landscape: Structural and mechanistic perspectives. Ann. Rev. Biochem. 2014, 83, 671–696. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Vermeulen, M. DNA methylation: Old dog, new tricks? Nat. Struct. Mol. Biol. 2014, 21, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.R.; Smith, E.; Shilatifard, A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007, 14, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- To, T.K.; Kim, J.-M.; Matsui, A.; Kurihara, Y.; Morosawa, T.; Ishida, J.; Tanaka, M.; Endo, T.; Kakutani, T.; Toyoda, T.; et al. Arabidopsis HDA6 regulates locus-directed heterochromatin silencing in cooperation with MET1. PLoS Genet. 2011, 7, e1002055. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Ann. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Saksouk, N.; Simboeck, E.; Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenet. Chromatin 2015, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sarnowska, E.; Gratkowska, D.M.; Sacharowski, S.P.; Cwiek, P.; Tohge, T.; Fernie, A.R.; Siedlecki, J.A.; Koncz, C.; Sarnowski, T.J. The role of SWI/SNF chromatin remodeling complexes in hormone crosstalk. Trends Plant Sci. 2016, 21, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Han, S.K.; Sang, Y.; Rodrigues, A.; Wu, M.F.; Rodriguez, P.L.; Wagner, D.; Biol, F. The SWI2/SNF2 chromatin remodeling ATPase BRAHMA represses abscisic acid responses in the absence of the stress stimulus in Arabidopsis. Plant Cell 2012, 24, 4892–48906. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.H.; Li, D.; Shimizu, H.; Nakamura, R.; Ishii, S. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 2011, 145, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Earley, K.; Lawrence, R.J.; Pontes, O.; Reuther, R.; Enciso, A.J.; Silva, M.; Neves, N.; Gross, M.; Viegas, W.; Pikaard, C.S. Erasure of histone acetylation by Arabidopsis HDA6 mediates large-scale gene silencing in nucleolar dominance. Genes Dev. 2006, 20, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Locatelli, S.; Varotto, S.; Donn, G.; Pirona, R.; Henderson, D.A.; Hartings, H.; Motto, M. Maize histone deacetylase hda101 is involved in plant development, gene transcription, and sequence-specific modulation of histone modification of genes and repeats. Plant Cell 2007, 19, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Grunstein, M. Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 2006, 7, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., III; Nishioka, K.; Reinberg, D. Histone lysine methylation: A signature for chromatin function. Trends Genet. 2003, 19, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, K.; Ding, Y.; Malkaram, S.; Riethoven, J.M.; Liu, R.; Yang, J.; Laczko, P.; Chen, H.; Xia, Y.; Ladunga, I.; et al. Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol. 2010, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Zhong, X.; You, J.; Xiong, L. Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol. Biol. 2013, 81, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Widiez, T.; Symeonidi, A.; Luo, C.; Lam, E.; Lawton, M.; Rensing, S.A. The chromatin landscape of the moss Physcomitrella patens and its dynamics during development and drought stress. Plant J. 2014, 79, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Muller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Blevins, T.; Pikaard, C.S. Arabidopsis histone lysine methyltransferases. Adv. Bot. Res. 2010, 53, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. PLoS ONE 2007, 2, e1210. [Google Scholar] [CrossRef] [PubMed]
- Camporeale, G.; Oommen, A.M.; Griffin, J.B.; Sarath, G.; Zempleni, J. K12-biotinylated histone H4 marks heterochromatin in human lymphoblastoma cells. J. Nutr. Biochem. 2007, 18, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013, 14, R59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Casas-Mollano, J.A.; Xu, J.; Riethoven, J.-J.M.; Zhang, C.; Cerutti, H. Osmotic stress induces phosphorylation of histone H3 at threonine 3 in pericentromeric regions of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2015, 112, 8487–8492. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ding, Y.; Sun, X.; Xie, S.; Wang, D.; Liu, X.; Su, L.; Wei, W.; Pan, L.; Zhou, D.X. Histone deacetylase HDA9 negatively regulates salt and drought stress responsiveness in Arabidopsis. J. Exp. Bot. 2016, 67, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Borsani, O.; Zhu, J.; Verslues, P.E.; Sunkar, R.; Zhu, J.K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 2005, 123, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Liu, X.; Thorn, G.; Duan, J.; Tian, L. Biochemical and biophysical research communications expression analysis of histone acetyltransferases in rice under drought stress. Biochem. Biophys. Res. Commun. 2014, 443, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Zaunbrecher, V.M.; Song, J.; Wendt, J.; Rosenbaum, H.; Madzima, T.H.; Sloan, A.E.; Huang, J.; et al. Genetic perturbation of the maize methylome. Plant Cell 2014, 26, 4602–4616. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-C.; Deng, B.; Liu, S.; Li, L.-M.; Hu, B.; Zhong, Y.-T.; Li, L. Isolation and characterization of an osmotic stress and ABA induced histone deacetylase in Arachis hypogaea. Front. Plant Sci. 2015, 6, 512. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luo, M.; Wang, Y.; Wu, K. Involvement of Arabidopsis histone deacetylase HDA6 in ABA and salt stress response. J. Exp. Bot. 2010, 61, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, X.; Singh, P.; Cui, Y.; Zimmerli, L.; Wu, K. Chromatin modifications and remodeling in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 129–136. [Google Scholar] [CrossRef] [PubMed]
- González, R.M.; Ricardi, M.M.; Iusem, N.D. Epigenetic marks in an adaptive water stress-responsive gene in tomato roots under normal and drought conditions. Epigenetics 2013, 8, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, M.; Bartucca, M.L.; Ciancaleoni, S.; Mimmo, T.; Cesco, S.; Pii, Y.; Albertini, E.; Del Buono, D. Iron deficiency in barley plants: Phytosiderophore release, iron translocation, and DNA methylation. Front. Plant Sci. 2015, 6, 514. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Sasaki, T.; Ueda, M.; Sako, K.; Seki, M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 2015, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; How-Kit, A.; Stammitti, L.; Teyssier, E.; Rolin, D.; Mortain-Bertrand, A.; Halle, S.; Liu, M.; Kong, J.; Wu, C.; et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. USA 2015, 112, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Burhans, W.C.; Curcio, M.J. Retrotransposition is associated with genome instability during chronological aging. Proc. Natl. Acad. Sci. USA 2011, 108, 20376–20381. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, A.; Tsementzi, D.; Tanriverdi, O.; Vlachonasios, K.E. Arabidopsis thaliana transcriptional co-activators ADA2 band SGF29a are implicated in salt stress responses. Planta 2011, 233, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Ji, D.; Li, S.; Wang, P.; Li, Q.; Xiang, F. The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS ONE 2012, 7, e41274. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Sharma, N.; Sahu, P.P.; Prasad, M. Chromatin-based epigenetic regulation of plant abiotic stress response. Curr. Genom. 2016, 17, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Epigenetic memory of stress responses in plants. J. Phytochem. Biochem. 2018, 2, e102. [Google Scholar]
- Verhoeven, K.J.F.; VanGurp, T.P. Transgenerational effects of stress exposure on offspring phenotypes in apomictic dandelion. PLoS ONE 2012, 7, e38605. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Wang, G.; Meyers, B.C.; Fellowship, D.Y. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2013, 2, e00354. [Google Scholar]
- Luna, E.; Bruce, T.J.A.; Roberts, M.R.; Flors, V.; Ton, J. Next-generation systemic acquired resistance. Plant Physiol. 2012, 158, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Ton, J. The epigenetic machinery controlling transgenerational systemic acquired resistance. Plant Signal. Behav. 2012, 7, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Havecker, E.R.; Wallbridge, L.M.; Fedito, P.; Hardcastle, T.J.; Baulcombe, D.C. Metastable differentially methylated regions within Arabidopsis inbred populations are associated with modified expression of non-coding transcripts. PLoS ONE 2012, 7, e45242. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Kim, E.D.; Ha, M.; Lackey, E.; Liu, J.; Zhang, Y.; Sun, Q.; Chen, Z.J. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009, 457, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, P.V.; Dunn, R.M.; Santos, B.A.; Bassett, A.; Baulcombe, D.C. Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J. 2012, 31, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, e124. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J. Inherited epigenetic variation—Revisiting soft inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Hauben, M.; Haesendonckx, B.; Standaert, E.; Van Der Kelen, K.; Azmi, A.; Akpo, H.; Van Breusegem, F.; Guisez, Y.; Bots, M.; Lambert, B.; et al. Energy use efficiency is characterized by an epigenetic component that can be directed. Proc. Natl. Acad. Sci. USA 2009, 106, 20109–20114. [Google Scholar] [CrossRef] [PubMed]
- Samota, M.K.; Sasi, M.; Awana, M.; Yadav, O.P.; Amitha Mithra, S.V.; Tyagi, A.; Kumar, S.; Singh, A. Elicitor-induced biochemical and molecular manifestations to improve drought tolerance in rice (Oryza sativa L.) through seed-priming. Front. Plant Sci. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Buck-Koehntop, B.A.; Defossez, P.A. On how mammalian transcription factors recognize methylated DNA. Epigenetics 2013, 8, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 1600. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Arul, L.; Talwar, D.; Raina, S.K. PCR amplification of minimal gene expression cassette: An alternative, low cost and easy approach to ‘clean DNA’ transformation. Curr. Sci. 2006, 91, 930–934. [Google Scholar]
- Kumar, S.; Chandra, A.; Pandey, K.C. Bacillus thuringiensis (Bt) transgenic crop: An environment friendly insect-pest management strategy. J. Environ. Biol. 2008, 29, 641–653. [Google Scholar] [PubMed]
- Stricker, S.H.; Köferle, A.; Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 2017, 18, 51–66. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S. Epigenomics of Plant Responses to Environmental Stress. Epigenomes 2018, 2, 6. https://doi.org/10.3390/epigenomes2010006
Kumar S. Epigenomics of Plant Responses to Environmental Stress. Epigenomes. 2018; 2(1):6. https://doi.org/10.3390/epigenomes2010006
Chicago/Turabian StyleKumar, Suresh. 2018. "Epigenomics of Plant Responses to Environmental Stress" Epigenomes 2, no. 1: 6. https://doi.org/10.3390/epigenomes2010006
APA StyleKumar, S. (2018). Epigenomics of Plant Responses to Environmental Stress. Epigenomes, 2(1), 6. https://doi.org/10.3390/epigenomes2010006